

 在线客服
在线客服
 快速发布
快速发布
 我的店铺
我的店铺
 我的化学加
我的化学加
 我的消息
我的消息
 我要充值
我要充值


 回到顶部
回到顶部



买产品

克难题

技术供需

求职招聘

发定制

项目整合

园区推荐

行业资讯

化学加智库

商家
买产品
克难题
技术供需
求职招聘
发定制
项目整合
园区推荐
行业资讯
化学加智库
商家
吡啶骨架是药物、材料及农药中出现频率最高的杂环之一,其中3-卤代吡啶因其独特的卤素效应可显著改善分子的代谢稳定性、脂溶性和蛋白结合能力,已成为上市药物(如尼麦角林、罗氟司特)及后续交叉偶联的关键合成子(图1a)。然而,由于吡啶环的π-亲核性较低且质子化倾向强,传统的亲电卤化反应要么仅能发生在富电子的吡啶环上,要么会“绕过”吡啶环,优先发生在电子云密度更高的苯环上(图1b)。基于上述挑战,研究者通常采用间接方法来实现吡啶间位的卤化反应。例如,McNally课题组利用开环的Zincke亚胺中间体实现了该转化,而Studer课题组则采用了类似的去芳构化-再芳构化策略(图1c)。然而,这些方法均存在合成步骤较多、反应条件复杂等局限性。
北京大学宋颂/焦宁研究团队一直致力于发展高效、绿色、经济的卤化方法,为药物分子的合成修饰及先导药物分子的发现提供新策略(Acc. Chem. Res.2024, 57, 3161),前期工作中,团队先后实现了富电子芳烃(Nat Catal.2020, 3, 107; Nat. Commun.2021, 12, 3873;CCS Chem.2020, 2, 566)、缺电子芳烃(J. Am. Chem. Soc.2022, 144, 13415;Chin. Chem. Lett.2025, 36, 111444)和吡啶对位的卤化修饰(Chem.2024, 10, 628)。基于以上工作积累,本研究利用溶剂化效应,克服了吡啶间位卤化在反应活性和区域选择性方面面临的难题,成功实现了吡啶的间位选择性卤化修饰反应。该方法条件简单温和,表现出优异的吡啶骨架选择性,解决了该领域近百年来的重大科学难题(图1d)。
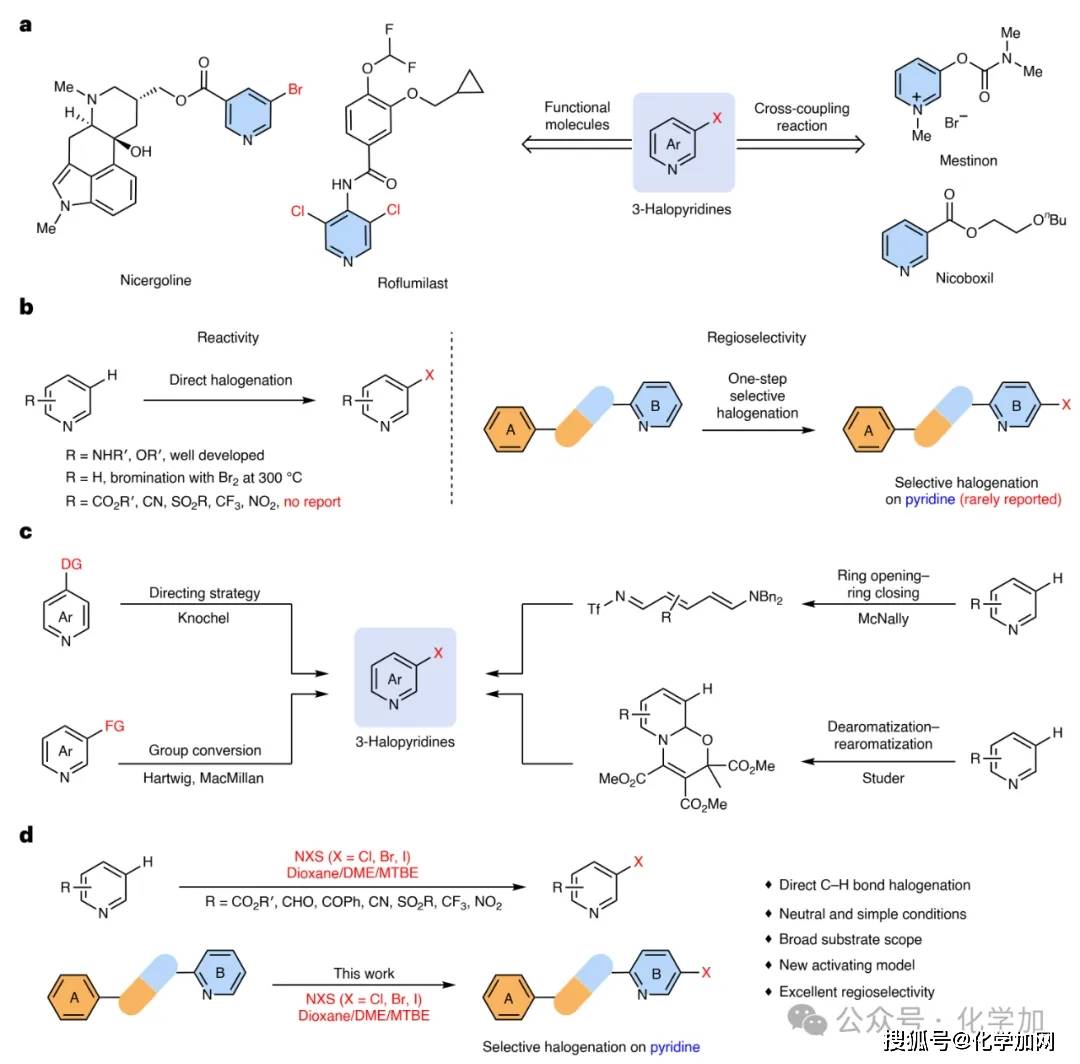
图1 研究背景
为攻克吸电子取代吡啶的卤化难题,本研究选取带有吸电子取代基的3-吡啶甲酸甲酯作为标准底物进行反应探索。令人欣喜的是,在二氧六环作为溶剂时,该反应表现出优异的反应性,能够以89%的收率获得间位溴化产物。其他醚类溶剂如四氢呋喃、甲基叔丁基醚等也可实现该转化,但收率略低。相比之下,非醚类溶剂(如六氟异丙醇、乙酸乙酯、甲苯、二甲基亚砜、乙腈、二氯乙烷等)均无法促进反应进行。进一步对溴源进行筛选,确定NBS为最优选择。此外,路易斯酸、布朗斯特酸及路易斯碱类催化剂均不能催化该反应。
在确定最优反应条件后,我们对吡啶间位溴化反应的底物适用性进行了系统评估(图2)。研究发现,含有酯基、氰基、硝基、三氟甲基、醛基、酮基、酰胺、磺胺及卤素等吸电子基团的吡啶均能良好兼容该反应体系,以30%–81%的收率获得相应的间位溴化产物。此外,苄基、烷基、氧苯基等给电子基取代的吡啶也能有效参与反应。杂环、芳基取代的吡啶同样表现良好,可获得中等至优良的收率。对于2位或4位取代吡啶,反应进一步生成双溴化产物;而2位吸电子基取代吡啶的反应性则相对较低。多取代吡啶亦展现出良好的反应兼容性。该策略还可拓展至喹啉、异喹啉和苯并喹啉等其他氮杂芳烃。通过调整醚类溶剂,该反应体系还可进一步实现吡啶间位选择性碘化与氯化反应。
基于上述结果,我们将该方法应用于多种药物及功能分子的后期修饰中,成功实现了对烟酸酯衍生物、中枢兴奋药尼可刹米、溴吡斯的明前体、血脂调节药依托贝特、抗心绞痛药物尼可地尔、肾上腺皮质激素美替拉酮、杀菌剂喹氧灵、抗基底细胞癌药物维莫德吉以及材料分子TmPyPB的卤化修饰。这些复杂分子中所含的酯基、酰胺、醚、酮等官能团在反应中均表现出良好的兼容性。上述结果充分证明,本方法在复杂分子的后期修饰中具有广泛的应用潜力,为药物研发提供了一条新颖有效的途径。
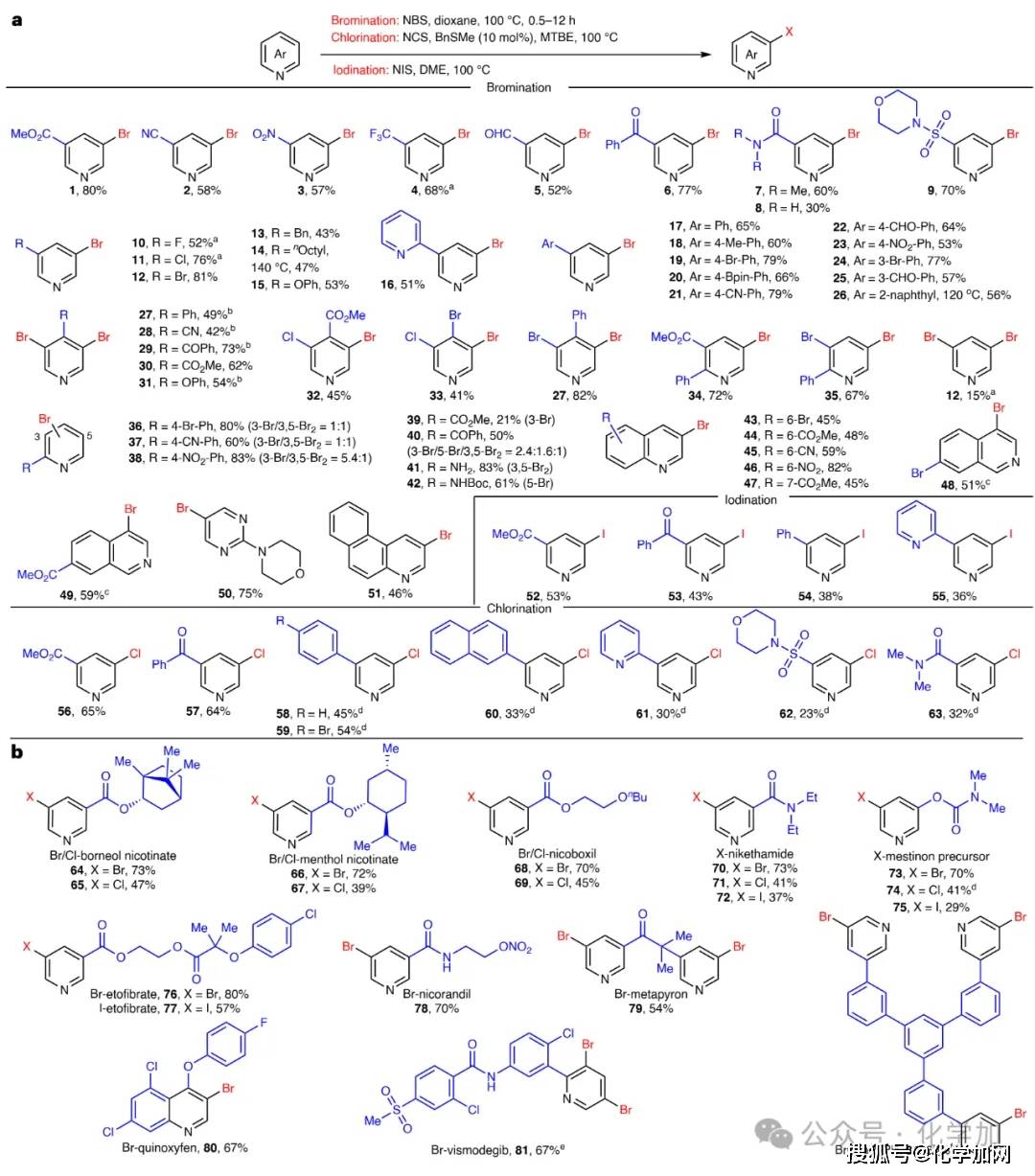
图2 吡啶间位卤化反应的底物范围
需要突出强调的是,本方法在保持高反应活性的基础上,成功解决了吡啶环相对于富电子芳环的选择性卤化这一关键挑战(图3)。在3-苯氧基吡啶、3-萘基吡啶、依托贝特、喹氧灵以及维莫德吉等同时含有富电子芳环与吡啶环的分子中,现有卤化体系如强质子酸体系(m-NBSA/HFIP, TfOH)、路易斯酸催化体系(AuCl3)以及路易斯碱催化体系(Trip-SMe)由于固有的电子效应倾向,主要生成芳环卤代产物。本方法则展现出独特的位点专一性,能够完全抑制富电子芳环的竞争性反应,精准实现吡啶环的间位卤化,并获得单一产物。

图3 已知方法与本方法的选择性对比
作为重要的合成子,间位卤代吡啶能够高效构筑更为复杂的分子结构(图4)。以其为起始原料,我们通过偶联反应成功实现了烷基化、烯基化、炔基化及膦酰化,充分展现了其在复杂结构构建中的关键作用。尤为值得一提的是,在克级规模放大实验中,该反应完成后无需柱色谱纯化,仅通过浓缩、石油醚洗涤与过滤,便能以71%的收率获得产物,凸显了其良好的实用性与可放大性。此外,该溴化反应还可与Suzuki-Miyaura偶联反应串联,无需分离卤代中间体,即可一步实现吡啶间位的直接芳基化。最后,我们还将该方法应用于药物尼麦角林的合成:从底物88出发,经酯化反应得到中间体89,随后在本体系条件下高选择性地实现了吡啶环的间位溴化,高效构筑了尼麦角林分子。
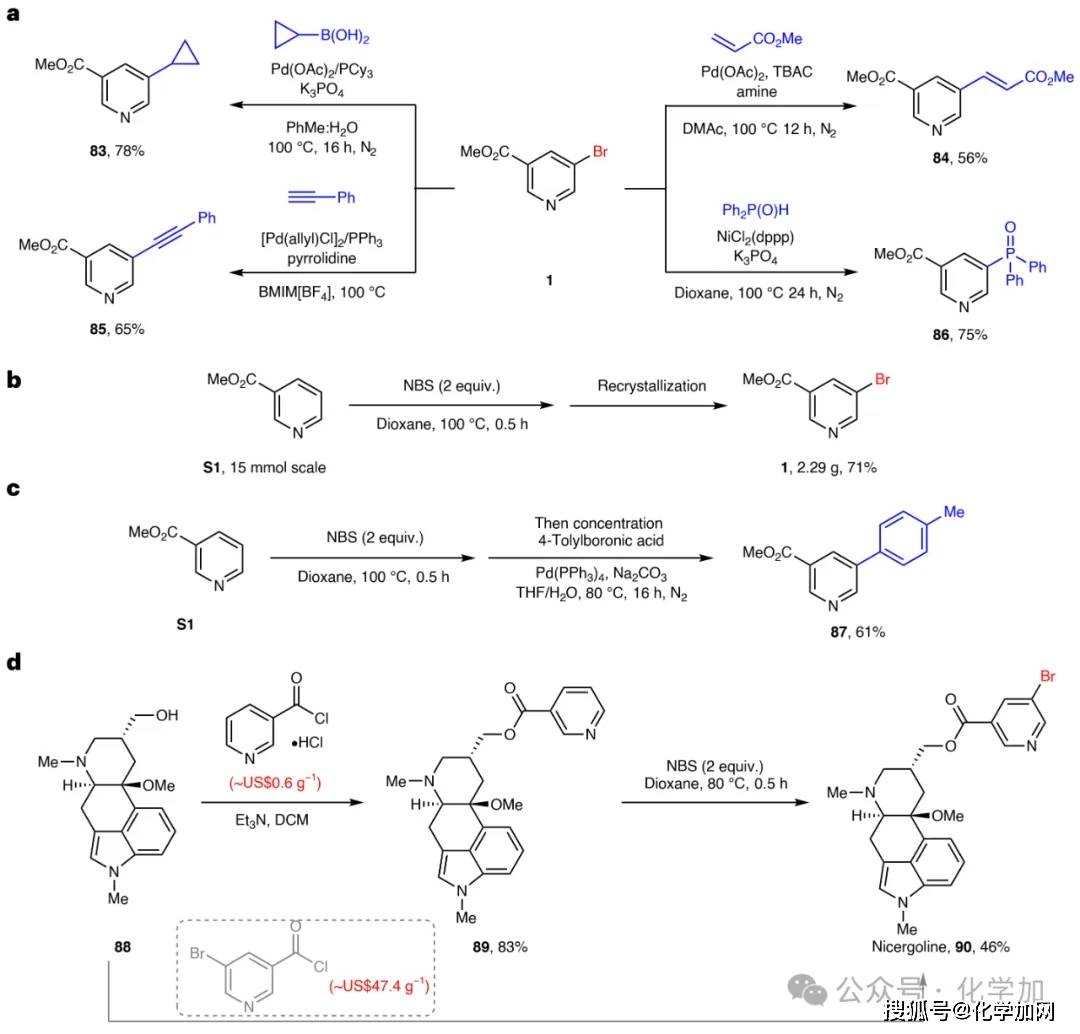
图4 间位卤代吡啶产物的应用
为阐明反应机理,该研究开展了一系列机理实验(图5)。初步的卤源筛选排除了溴单质或亲核溴源(如NaBr、TBAB)作为活性溴物种的可能,黑暗实验则排除了光促反应路径。自由基捕获实验(TEMPO抑制、1,1-二苯基乙烯溴化)揭示了反应经历自由基过程,且吡啶的存在是产生溴自由基的必要条件。底物结构-活性关系研究表明,吡啶氮原子是反应发生的必备位点:其质子化或2,6-位取代均导致反应完全抑制,而苯甲酸甲酯类似物也无反应活性。异喹啉S49与NBS反应意外生成胺化产物95,进一步暗示胺化类似物可能为反应的中间体。¹³C-NMR研究显示吡啶及醚类溶剂的加入均使NBS分子中羰基碳信号明显低场位移,而琥珀酰亚胺羰基信号保持不变,证实二者作为路易斯碱分别与NBS的溴原子发生相互作用,共同促进溴-氮键均裂产生溴自由基,从而驱动了整个自由基卤化过程。
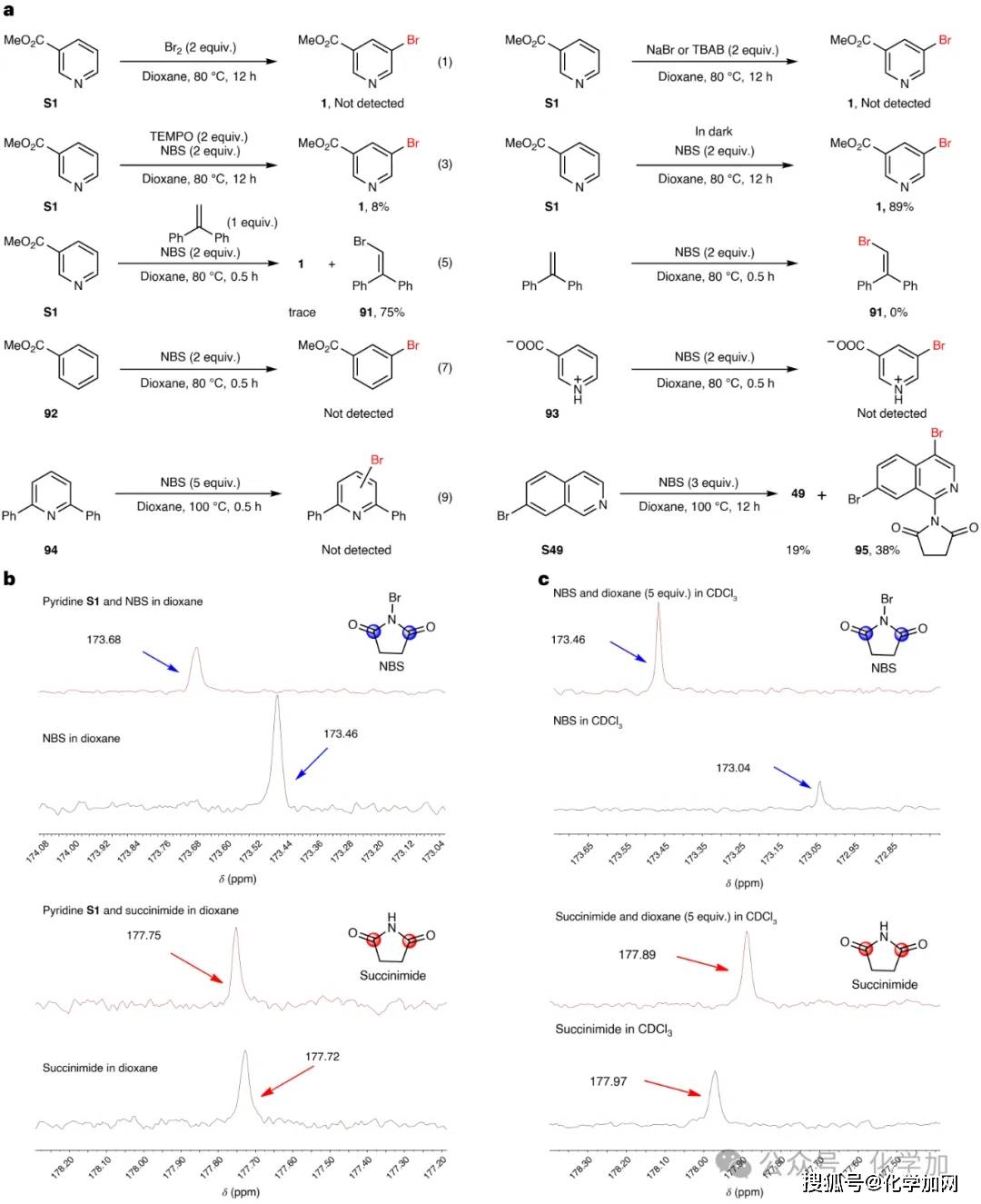
图5 机理实验
与上海大学郦鑫耀教授合作通过DFT计算研究了二氧六环中吡啶C-3位卤代反应的机理(图6)。卤代试剂受热产生不稳定的Br⁺和Br∙,两者均可被吡啶和二氧六环稳定化。反应始于吡啶与NBS形成复合物,并生成关键的吡啶-溴自由基中间体,然后进一步转化成Py-Br-NS(C)中间体,其结构得到HRMS验证。紧接着溴自由基加成到该中间体,倾向于进攻C-3位。最后去质子化步骤有两条路径,其中涉及溴离子参与的路径能垒最低,反应路径最优,最终芳构化得到溴化产物。前线轨道分析表明,关键中间体Py-Br-NS(C)的HOMO与溴自由基的SOMO能差小,从而解释了其高反应活性。
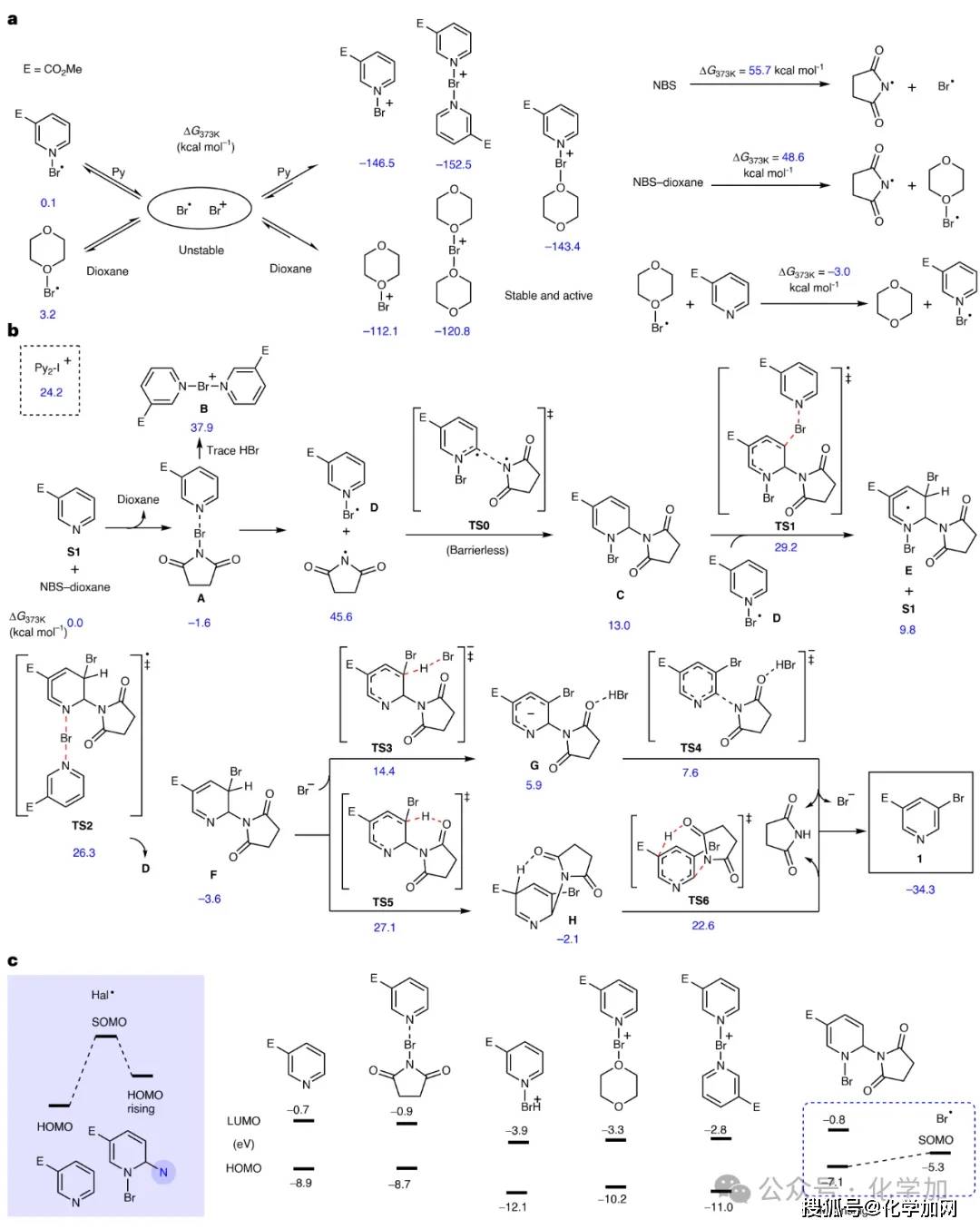
图6 DFT计算
总结
综上所述,该团队利用廉价醚类溶剂的溶剂化效应,实现了直接的吡啶间位选择性卤化反应。本方法条件简单温和,能够兼容各种吸电子取代基并表现出优异的吡啶骨架选择性,解决了这一领域近百年以来的重大科学难题。机理实验表明该反应经历自由基过程,“醚溶剂-吡啶-卤源”的协同活化是反应的关键,1-卤-2-胺二氢吡啶可能是反应的中间体。
宋颂研究员和焦宁教授为本文的共同通讯作者,北京大学药学院博士后李超,上海大学郦鑫耀教授和北京大学药学院六年制学生李佳星为本论文的共同第一作者。北京大学药学院六年制学生王之行,欧阳敦煌为共同作者。北京大学药学院天然药物及仿生药物全国重点实验室为第一通讯单位,细胞稳态与衰老性重大疾病北京研究中心为共同通讯单位。该研究得到国家自然科学基金委、国家重点研发计划、北大医学顶尖学科及学科群发展专项等项目的支持。
文献详情:
Li, C., Li, X., Li, J. et al. Direct regioselective C-3 halogenation of pyridines. Nat. Synth (2025).
https://doi.org/10.1038/s44160-025-00915-3 s声明:化学加刊发或者转载此文只是出于传递、分享更多信息之目的,并不意味认同其观点或证实其描述。若有来源标注错误或侵犯了您的合法权益,请作者持权属证明与本网联系,我们将及时更正、删除,谢谢。 电话:18676881059,邮箱:gongjian@huaxuejia.cn
