

 在线客服
在线客服
 快速发布
快速发布
 我的店铺
我的店铺
 我的化学加
我的化学加
 我的消息
我的消息
 我要充值
我要充值


 回到顶部
回到顶部



买产品

克难题

技术供需

发定制

项目整合

园区推荐

行业资讯

化学加智库

商家
买产品
克难题
技术供需
发定制
项目整合
园区推荐
行业资讯
化学加智库
商家
为特定合成应用确定最佳手性催化剂结构是对映选择性合成的核心挑战,通常需要进行大量经验性的、迭代的优化。这些工作受益于“特权”手性催化剂的存在,它们通常是催化剂精细化的良好起点(图1A)。由于不对称反应优化通常需要考察许多催化剂类似物,最常见的特权催化剂框架是模块化的,能够微调手性环境并生成用于筛选的催化剂类似物库。当催化剂可以从市售的对映体纯原料(“手性源”)以最少的步骤合成时,这一点尤其实用。
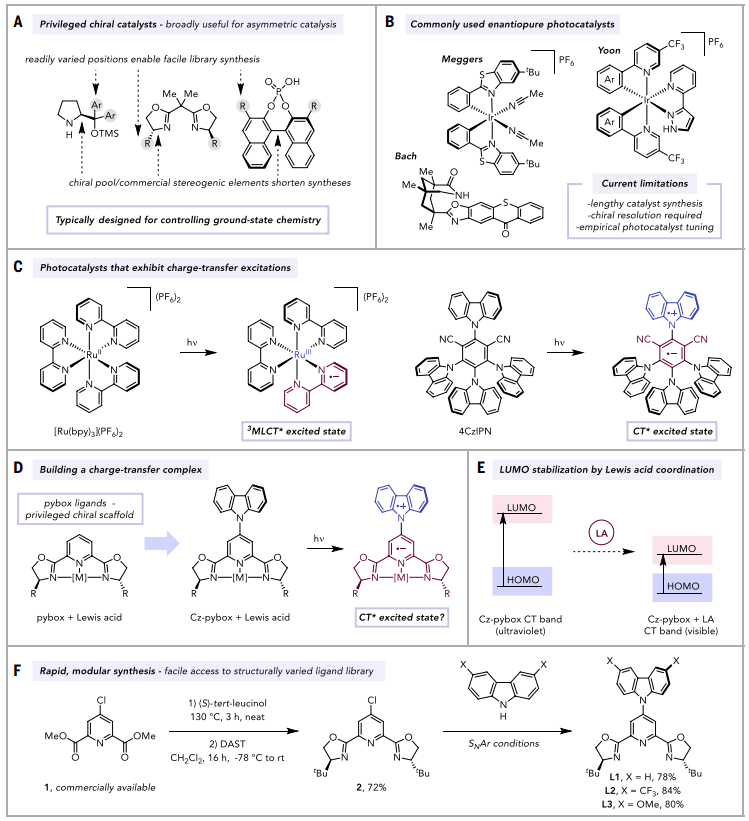
图1. 特权光催化剂的设计(图片来源:Science)
不对称光催化中,高效且通用的手性光催化剂非常稀缺。大多数现有光催化剂合成路线长、需要手性拆分,且适用范围有限。虽然有一些“特权结构”催化剂(如 bis(oxazoline)、BINOL磷酸等)在基态反应中表现优异,但它们在光催化中应用受限(图1B)。开发一类合成上易得、结构可调、机制上通用的特权手性光催化剂,可以促进其更广泛领域的应用,并极大地加速不对称光化学的进展。
吡啶双(噁唑啉)配合物是最被深入理解的特权催化剂结构之一,其中心吡啶配体作者设想可作为CT(charge-transfer,电荷转移)光催化剂的受体部分。作者认为,在吡啶的4-位引入咔唑单元,将在咔唑为中心的最高占据分子轨道和吡啶为中心的最低未占分子轨道之间产生一个可及的CT跃迁。此外,类似于吖啶、咔唑基联吡啶和其他给体-受体系统的路易斯酸配合物,预期吡啶基团与路易斯酸的配位将稳定LUMO,这将转化为更低的S1态能量,理想情况下可达可见光激发范围。类似的给体-受体pybox配合物先前已被合成用于圆偏振发光应用,但此类配合物尚未在光催化背景下被探索。
作为对设计策略的初步测试,作者以合成Cz-tBu-pybox配体L1为目标。已知的氯化前体2可以通过成熟的两步一锅法程序轻松制备,在常规的亲核芳香取代条件下,咔唑单元可以高产率地安装上去。这种合成方法具有高度模块化:手性噁唑啉定义的立体化学环境可以通过使用市售的各种对映体纯氨基醇开始合成来改变,而配体的电子性质可以使用SNAr步骤中不同的N-杂环给体轻松调节。因为配体的立体化学信息来源于手性源,所以该配体可以通过简单的柱层析纯化,无需手性拆分。
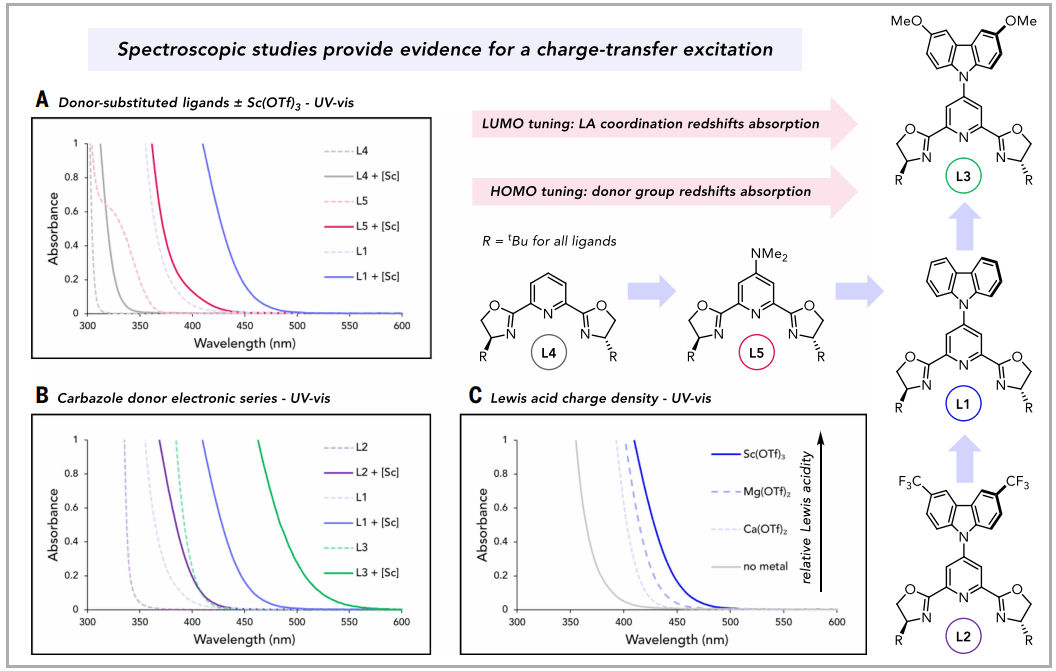
图2. 紫外-可见吸收光谱(图片来源:Science)
接下来,使用紫外-可见吸收光谱验证了本文的CT设计原理(图2)。随着取代基从H变为NMe2再变为Cz,吸收光谱的起始逐渐发生红移;然而,这些自由配体在400 nm以上都没有强吸收。加入Sc(OTf)3作为代表性路易斯酸,导致每种配体的UV-vis吸收发生红移,使L1能够吸收可见光。与这些数据一致,加入Sc(OTf)3后,L1的溶液颜色从无色变为黄色。与母体配体L1和配合物[Sc(Cz-tBu-pybox)(OTf)3]相比,在Cz的3-位和8-位用吸电子CF3基团或给电子OMe基团取代,会使吸收发生蓝移或红移。这意味着HOMO能级被选择性调节,并且低能量吸收带的性质确实是CT。这些结构-性能关系趋势得到了密度泛函理论计算的支持。该系列配体的稳态发射光谱反映了激发态能量的相同趋势。
在DCM中对配体和配合物进行的时间分辨光致发光测量显示了多指数衰减动力学,配合物的最长寿命组分在7到17纳秒之间,而配体的最长寿命组分往往稍快。最小的ΔEST为0.18 eV,对应[Sc((CF3)2Cz-tBu-pybox)(OTf)3],其在DCM中的激发态寿命是该族配合物中最长的。
接下来,在系列模型不对称光反应中评估了这些光催化剂的潜力。每个模型反应先前都是在双催化条件下报道的,使用手性Sc/pybox催化剂与消旋的Ru基或Ir基光催化剂串联。最初,考察了由α-硅基苯胺的光催化氧化引发的对映选择性Giese型α-氨基自由基加成反应。使用[Sc(Cz-iBu-pybox)(OTf)₃]同时作为光催化剂和立体诱导源,确定了反应条件,在此条件下可以生成Giese加成产物5a,其产率和ee值与原报道完全相同。在所有情况下,都观察到相似的高产率和对映选择性。这些结果表明,咔唑单元的引入并没有实质性地扰乱Sc(III)中心周围的手性环境。事实上,改变Cz-pybox骨架上噁唑啉的取代基,导致了在双催化剂系统中观察到的相同的对映选择性趋势,由亮氨醇衍生的iBu噁唑啉在两种系统中都提供了最优的ee值(图3)。
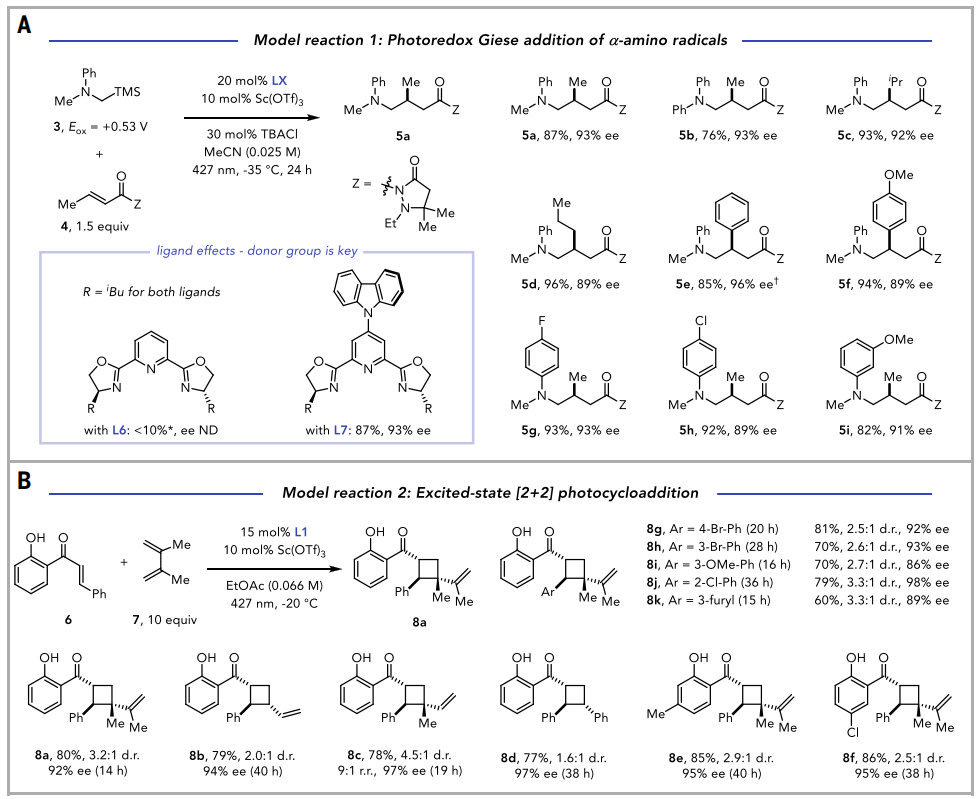
图3. 对映选择性应用(图片来源:Science)
在确立了[Sc(Cz-iBu-pybox)(OTf)3]作为不对称光氧化还原催化剂的有效性后,接下来考察了其在不对称光诱导能量转移反应中的性能。选择了一个2'-羟基查尔酮6的模型[2+2]光环加成反应,与光氧化触发的Giese加成一样,也能够确定反应条件,在该条件下形成环丁烷产物8,其产率、立体选择性和普适性几乎与原报道相同,这表明[Sc(Cz-iBu-pybox)(OTf)3]可以同时充当手性路易斯酸催化剂和三重态敏化剂的角色。在这些研究中,双催化剂系统中最佳的叔亮氨醇衍生的噁唑啉取代基在Cz-pybox骨架上也被证明是最理想的。即使反应时间延长,反应也可以在低负载量的手性催化剂下进行,这与单催化剂系统相对于双催化方法在动力学上具有优势的假设一致。这些结果表明,在先前应用中广泛使用pybox配体所获得的经验知识,可以很容易地转化到由这个光催化剂家族介导的光氧化还原和激发态反应中。
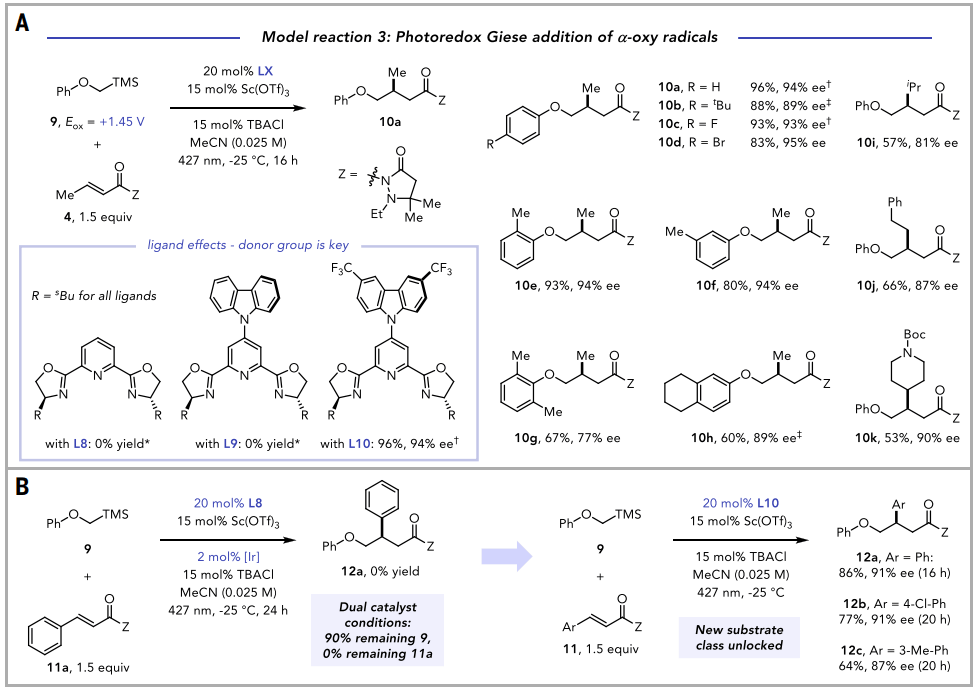
图4. Giese 加成反应的其他应用(图片来源:Science)
双催化不对称光反应的一个重要策略特征是能够改变光催化剂结构,以合理优化热力学参数,如三重态能量和氧化还原电位。对配体L1-L3与Sc(OTf)3的1:1混合物进行的循环伏安研究表明,Eox值呈现出直观的趋势。尽管引入这些不同的咔唑给体基团显著改变了相应Sc配合物的光物理和电化学性质,但通过三苯基氧膦路易斯酸探针的31P核磁谱判断,这些配合物具有几乎相同的路易斯酸度。
修改催化剂电化学性质的能力在将此类光催化剂应用于对映选择性α-氧基自由基Giese加成反应中被证明至关重要。尽管这个反应与上述α-氨基自由基加成反应表面上相似,但α-硅基醚9比α-硅基胺3更难氧化。与这个反应需要更强的氧化电位一致,作者发现[Sc(Cz-iBu-pybox)(OTf)3]不是此转化的有效光氧化还原催化剂。然而,含有吸电子三氟甲基基团的催化剂[Sc((CF3)2Cz- iBu-pybox)(OTf)3]由于其对自由基前体和Giese受体都表现出相似的底物普适性,被证明在此Giese反应中非常有效,在改变原体系其他反应参数后,母体Giese加成产物以高产率和优异的对映选择性获得(图4)。
尽管本文探讨的所有三个模型反应都可以用双催化剂系统完成,但在许多情况下,使用手性光催化剂可以解锁新的反应性或选择性。使用Cz-pybox平台能够以高产率和对映选择性合成多种先前难以获得的芳基取代的Giese加合物。这种反应性的差异可能是由于缺电子咔唑光催化剂的高度氧化性,导致这两种光催化剂的激发态在能量转移和电子转移过程之间分配不同。除了确立Cz-pybox光催化剂替代双催化剂系统的能力,这些结果也突显了该平台在开发独特的不对称光反应方面的有效性。
作者开发的手性光催化剂平台已经具备了成熟的、高度通用的特权催化剂系统的几个关键特征。首先,从市售手性源材料出发,Cz-pybox配体的简短、模块化合成,能够快速合成具有多个可调特征的催化剂类似物。第二,手性路易斯酸配合物的光电性质很容易被理解为源于可及的给体-受体电荷转移态,这有助于对其进行合理调控,以优化机制不同的光反应。最后,由于这种光催化剂设计利用了pybox配合物的核心结构特征,而pybox配合物是已知的最被深入理解和多功能的特权催化剂之一,预期这些手性光活性路易斯酸将展现出类似的一般性能力,能够控制机制多样的光催化反应的对映体决定态。
Riley M. Kelch, Lea Hämmerling, Eli Zysman-Colman*, Tehshik P. Yoon*. Modular enantioselective photocatalysts from privileged pybox scaffolds. Science, 2026,doi:10.1126/science.aeb5832

声明:化学加刊发或者转载此文只是出于传递、分享更多信息之目的,并不意味认同其观点或证实其描述。若有来源标注错误或侵犯了您的合法权益,请作者持权属证明与本网联系,我们将及时更正、删除,谢谢。 电话:18676881059,邮箱:gongjian@huaxuejia.cn
