

 在线客服
在线客服
 快速发布
快速发布
 我的店铺
我的店铺
 我的化学加
我的化学加
 我的消息
我的消息
 我要充值
我要充值


 回到顶部
回到顶部



买产品

克难题

技术供需

发定制

项目整合

园区推荐

行业资讯

化学加智库

商家
买产品
克难题
技术供需
发定制
项目整合
园区推荐
行业资讯
化学加智库
商家

目前的商用锂离子电池皆是基于锂离子在正负极之间的可逆嵌入、脱出反应,电解液为EC基的酯基电解液。经过20余年的发展与优化,其能量密度已经接近其理论值。要进一步提高其能量密度,必然要开发新的正极材料、负极材料以及与之相匹配的电解液体系。在所有的可能的负极材料中,Li金属具有最高的理论容量 (3860 mAh/g),最低的电位 (-3.04 V vs SHE),故最近几年受到了广泛关注。而与此同时,正极材料一直是锂离子电池能量密度进一步提升的关键性限制因素。因此,要大幅提高锂离子电池的能量密度,在利用锂金属替代目前的嵌入式石墨负极材料的同时,必须要利用更高能量密度的正极材料替代现有的LiCoO2、LiFePO4等正极材料。
目前为止,由于锂离子电池宽的正负极电压窗口,没有任何一种电解液能够同时对锂离子电池的高压正极材料和低压负极材料同时热力学稳定。故形成有效的CEI/SEI膜是实现正负极可逆循环的关键因素。EC基电解液由于能够在石墨负极一侧形成稳定的SEI,而同时其正极一侧的稳定窗口可以达到4.3 V左右,故被广泛的应用于目前的基于石墨的锂离子电池材料。而EC酯基电解液,对锂沉积溶解的库伦效率仅为80-90%,而正极一侧的稳定窗口也偏低,限制了其在新一代更高能量密度电池中的应用。醚基电解液由于其耐还原,虽然对锂金属具有较高的库伦效率,但其正极一侧稳定窗口比EC酯基电解液还低。故要进一步提高能量密度,必然要开发新的电解液以匹配更高电压的正极材料,同时也能匹配锂金属负极材料。
【图文解析】
图1为不同电解液的电化学性能对比。与传统的酯基电解液相比,1M LiPF6FEC/FEMC/HFE表现出了更高的Li金属沉积-溶解库伦效率。在0.2 mA/cm2的电流下,经过50 周期循环后,库伦效率逐渐提升至99.2%左右。而对于DMC电解液,库伦效率仅为20-30%,加入EC后,库伦效率可以提升至80%左右。将EC替换成FEC,库伦效率可以进一步提升至96%左右。更为显著的提升为高压正极一侧的稳定性,对于EC/DMC体系,从4.3V开始出现氧化峰。将EC替换成FEC,稳定性有较大改善,但当电位提升到4.8 V左右,依然开始出现较大的氧化电流。而对于1M LiPF6FEC/FEMC/HFE电解液,电位提升至5.8 V左右,其氧化电位依然较小。上述表明1M LiPF6 FEC/FEMC/HFE电解液具有良好的正极稳定性,同时大幅改善了Li金属负极可逆沉积-溶解效率。
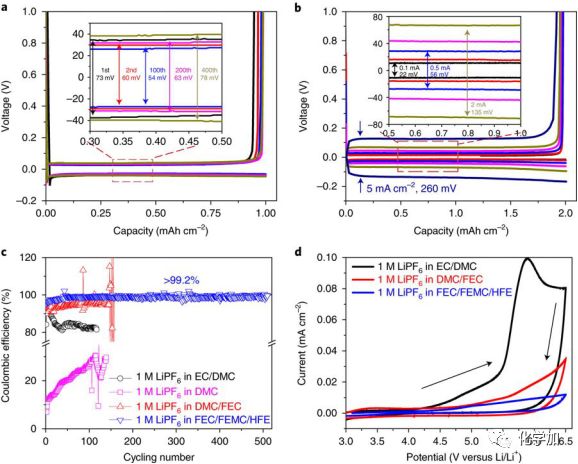
图1. 不同电解液的电化学性能对比。(a)Li金属在1M LiPF6FEC/FEMC/HFE氟化电解液中的沉积-溶解曲线;(b)不同沉积-溶解电流下的曲线;(c)不同电解液的Li金属的沉积-溶解库伦效率对比,电流密度0.2 mA/cm2;(d)不同电解液的氧化CV曲线。扫描速度:5 mV/s.
在不同电解液中循环后,锂金属表现出了截然不同的形貌,如下图2所示。在普通的EC/DMC酯基电解液中,大量的锂枝晶产生。此锂枝晶的生成,大幅增加了比表面积,降低了锂金属负极的体积容量密度。并且锂枝晶的生成容易形成死锂、穿透隔膜引起安全问题。经过多次循环后,锂金属的厚度也大幅增加。利用FEC替换EC后,可以大幅抑制锂枝晶的生成,但其表面积仍有大幅增加。当电解液换成1M LiPF6FEC/FEMC/HFE后,锂枝晶完全被抑制,锂金属以圆球状的形态出现。此形貌的产生应该与大量生成的以氟化锂为主的无机SEI有关。

图2.不同电解液中锂金属循环后的SEM形貌。(a-c)1M LiPF6in EC/DMC; (d-f): 1M LiPF6 in FEC/DMC; (g-i) 1 M LiPF6in FEC/FEMC/HFE.
以NMC811(4.4 V)和LCP(5 V)为正极,我们配成了相应的锂金属电池, 1 M LiPF6in FEC/FEMC/HFE电解液大幅提高了其循环稳定性和库伦效率。对于NMC811,循环450周期后,容量保持率为90%左右。而对于LiCoPO4,循环1000周期后,其容量保持率高达93%左右,并且与1 M LiPF6in FEC/DMC相比,其库伦效率有了大幅提高,从95%提升到99.8%以上。并且充电到5V后,静置48小时后,LiCoPO4依然可以放出91.6%的容量,而对于1 M LiPF6in FEC/DMC,仅能放出37.9%的容量。
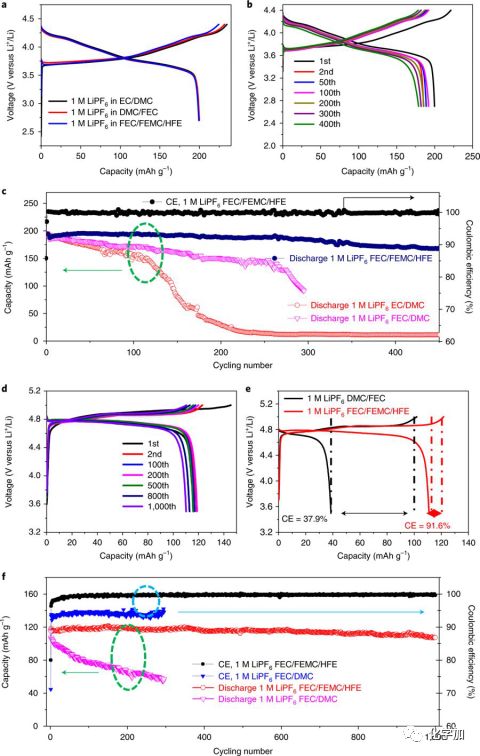
图3. 以NMC811和LiCoPO4为正极的锂金属电池的电化学性能。 (a)Li||NMC811首次充放电曲线;(b)Li||NMC811在1M LiPF6FEC/FEMC/HFE电解液中不同循环的充放电曲线;(c)Li||NMC811电池在不同电解液中的循环性能对比;(d)Li||LiCoPO4在1M LiPF6FEC/FEMC/HFE电解液中不同循环的充放电曲线;(e)Li||LiCoPO4电池充电后,静置48小时后,放电容量及其效率;(f)Li||LiCoPO4电池在不同电解液中的循环稳定性对比。
理论计算表明1M LiPF6FEC/FEMC/HFE电解液中的几种溶剂在较高电位下(1.5 V-2.5 V)即可通过脱氟分解,溶剂脱下的氟在电解液中极易与Li+结合,进而生成以LiF为主的SEI。Ar+刻蚀表明Li金属在1M LiPF6FEC/FEMC/HFE电解液中循环后的SEI在LiF的含量高达90%以上。而正极一侧,在通过溶剂脱氢反应,之后生成富含氟的复合物CEI (如图5所示)
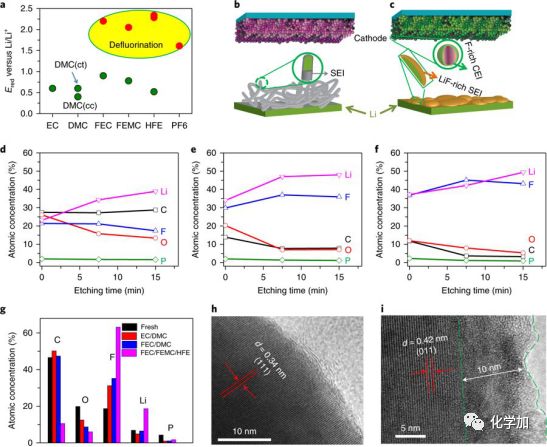
图4. 电解液不同溶剂的分解过程理论计算及其在正负极材料上的界面分析。(a)电解液中溶剂化的溶剂及PF6阴离子分解电位计算(G4MP2 calculations with the SMD implicit solvent model; (b、c) 不同电解液中形成的锂金属负极SEI和正极CEI对比示意图,b普通电解液,c 1M LiPF6FEC/FEMC/HFE电解液。(d-f)不同电解液中Li金属表面的SEI成分随Ar+刻蚀时间变化,d 1 M LiPF6EC/DMC, e LiPF6 FEC/DMC, f 1 M LiPF6FEC/FEMC/HFE. (g)LiCoPO4正极材料在不同电解液中循环后的CEI成分对比。(h)原始的LiCoPO4正极材料的高分辨透射照片;(i)在1 M LiPF6 FEC/FEMC/HFE电解液中循环后的LiCoPO4正极材料的高分辨透射照片。
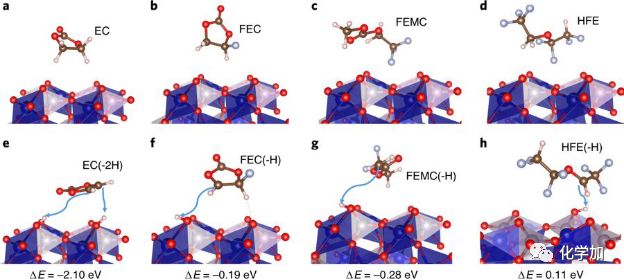
图5. 几种不同溶剂在充电状态的CoPO4正极表面的理论计算 (PBE+UDFT)的可能反应。(a-d)初始状态;(e-h)反应状态及其相应的反应能量。
在这项工作中,研究人员使用锂金属阳极和高容量/高压阴极开发了一种不易燃的全氟化电解质,用于腐蚀性电池化学。该电解质同时解决了这种高能电池面临的四个最紧迫的挑战:(1)不良的镀锂/剥离;(2)脱锂阴极表面的电解质氧化; (3)锂枝晶形成;;(4)安全性差。这项工作中选择的两种阴极化学物质(NMC811,LCP)仅具有代表性,因为它们具有有利的性质(具有强催化富Ni表面的高容量和异常高的电压)而被选择,并且在中间显示了相间氟化的概念。目前的工作应普遍适用于其他电池化学品,如钠金属。事实上,还观察到Na电镀/剥离的显着增加和氧化稳定性的改善,该研究结果为高盐浓度方法提供了另一种途径,用于设计用于最具侵蚀性的阴极和金属阳极化学品的新电解质系统。
参考文献
Xiulin Fan, Long Chen, Oleg Borodin, Xiao Ji, Ji Chen, Singyuk Hou, Tao Deng, Jing Zheng, Chongyin Yang, Sz-Chian Liou, Khalil Amine, Kang Xu & Chunsheng Wang . Non-flammable electrolyte enables Li-metal batteries with aggressive cathode chemistries.
Nature Nanotechnology, 2018, https://www.nature.com/articles/s41565-018-0183-2
声明:化学加刊发或者转载此文只是出于传递、分享更多信息之目的,并不意味认同其观点或证实其描述。若有来源标注错误或侵犯了您的合法权益,请作者持权属证明与本网联系,我们将及时更正、删除,谢谢。 电话:18676881059,邮箱:gongjian@huaxuejia.cn
