

 在线客服
在线客服
 快速发布
快速发布
 我的店铺
我的店铺
 我的化学加
我的化学加
 我的消息
我的消息
 我要充值
我要充值


 回到顶部
回到顶部













近日,西湖大学石航教授课题组在JACS上报道了一种铑催化的苯酚胺化,提供了各种苯胺的简洁合成,其中水是唯一的副产物。亲芳铑催化剂通过π-配位促进苯酚固有的困难的酮-烯醇互变异构化,允许随后与胺的脱水缩合。作者通过对具有各种电性的大量酚类和多种伯胺和仲胺进行反应,证明了这种氧化还原中性催化的普适性。结构复杂的生物活性分子(包括药物)的后期功能化进一步说明了该方法潜在的广泛用途。文章DOI:10.1021/jacs.1c12622
苯胺是功能性材料、医药、农产品和天然产物中常见的结构单元,因此,开发用于制备具有苯胺结构单元的化合物的有效方法是合成化学中的一项重要工作。在过去的几十年中,使用钯、铜或镍催化剂,在过渡金属催化的芳基卤化物和芳基硼酸等芳香族化合物的胺化领域取得了长足的进步。然而,酚的胺化仍然难以捉摸。由于Ph-OH键的高解离能(> 450 kJ/mol)、酚羟基对氧化等转化的敏感性以及它们对过渡金属质子化的倾向,酚类不适合过渡金属-催化的交叉偶联反应。在过渡金属催化的C-N键形成反应(包括Buchwald-Hartwig胺化和Ni-催化的胺化)中,三氟甲磺酰基等活化单元对于促进C-O键的氧化加成是必不可少的(图1a)。下载化学加APP,阅读更有效率。
图1. 从苯酚合成苯胺(图片来源:J. Am. Chem. Soc.)
在制定苯胺的简明合成策略时,作者借鉴了广泛应用于有机催化、还原胺化和Mannich反应的羰基胺缩合反应;他们的想法是,如果缩合可以与苯酚固有的困难的酮-烯醇互变异构化相结合,就可以开发出直接获得苯胺的方法。互变异构平衡取决于酮和烯醇互变异构体的相对稳定性(图1b)。对于丙酮,在正常条件下,平衡强烈有利于酮的形成,而苯酚的二烯酮互变异构体的形成由于失去芳香性而被完全抑制。1904年,Bucherer报道了亚硫酸氢钠促进的萘酚胺化,其中萘酚可以互变异构为烯酮,因为稠环去芳构化的能垒相对较低。尽管苯胺可以通过使用非均相催化剂的优雅氢化去芳构化/脱氢芳构化序列从酚类和胺类中生成,但外部或内部氢源是必不可少的。此外,苯酚与胺的胺化可以通过一系列氧化去芳构化、缩合和氧化还原异构化来完成。
鉴于Lewis酸可以推动酮-烯醇平衡向烯醇化物方向发展,作者想知道酸是否会特异性地激活酚环以迫使形成二烯酮而不是酚盐,从而为与胺的脱水缩合提供机会。在以往对金属-芳烃π-配位配合物的研究中,发现η6-苯酚配合物由于金属的吸电子作用容易去质子化,生成酮形式的η5-苯氧基配合物。然而,这种反应在催化中的应用仍有待证明。
在此,作者报道了一种无需活化试剂或还原剂/氧化剂的铑催化酚与胺直接胺化的有效方法(图1c)。该方法在原子经济性、酚类可再生性、以及废弃物等方面可以满足“绿色”。作者设想这种氧化还原中性催化将通过以下机制进行。首先,亲芳性的Rh(III)催化剂会通过π-配位与苯酚连接,去质子化后产生瞬时的η5-苯氧基配合物II;II的羰基胺缩合得到亚胺中间体III,它会发生互变异构,然后进行芳烃解离以产生所需的苯胺产物。
首先,作者以4-甲基苯酚(1a)和哌啶(2a)之间的模型反应开始了研究(图2a)。通过广泛的条件筛选,发现 [Cp*RhCl2]2(催化剂1)在含有10 mol% Na2CO3的庚烷中,在120 °C下得到所需的产物3a,收率为20%。将Cp*环上的一个甲基替换为氢(催化剂2)或脂肪族基团,如环己基(催化剂3)、异丙基(催化剂4)或三氟甲基(催化剂5),降低了产率。相反,具有芳族取代基的催化剂(6-10)的产率与使用催化剂1的产率相似或更好。特别是10,具有3,5-二(三氟甲基)-苯基,产率为40%。将反应温度提高到140 ℃,收率提高到80%,加入分子筛使收率提高到91%。使用预先形成的铑单体[CpCF3Rh(MeCN)3]2+ (催化剂11),在不需要 Na2CO3的情况下得到70%的3a。
有了最佳条件,作者探索了苯酚与哌啶2a的反应范围(图2b)。广泛的对位取代苯酚是合适的,以高达90%的产率提供所需的产物3b-3y。电中性(3b-3l)、给电子(3m-3p)和吸电子(3q-3t)基团和芳香环(3u-3y)均耐受。无论取代物(3z-3ae)的电性如何,一系列间位取代的苯酚都经历了所需的反应。此外,多取代苯酚(3af-3ah)和稠环苯酚(3ai-3ao)的胺化反应以中等至良好的收率平稳地提供苯胺。值得注意的是,杂环部分,包括吡啶(3y)、二噁英(3ak)和四氢喹啉(3am和3an),在反应条件下都是可耐受的。胺化对羟基的空间位阻敏感,2-甲基苯酚仅产生痕量所需产物(3ap)。
图2. 条件筛选及苯酚的底物范围(图片来源:J. Am. Chem. Soc.)
接下来,作者通过与4-甲基苯酚(1a)进行反应来评估胺的反应范围(图3)。各种带有直链(3aq-3ax)、β-支链(3ay-3bc)或α-支链(3bd-3bi)取代基的伯胺都是合适的,以40-89%的产率提供相应的苯胺。此外,苄胺(3bk)和各种衍生物(3bl-3bp)在贵金属存在下易于β-H消除,也与反应条件相容。本文的方法也适用于广泛的仲胺。具体来说,药物中常见的哌啶(3bq-3cd)和哌嗪(3ce-3cl)是优良的底物,以及一系列反应性官能团,包括腈(3bq)、羧酸酯(3bs)、1°-3 °醇(3bt,3bu,3by),氨基(3bv,3ce-3cg),酰胺(3cd),氨基甲酸酯(3bw,3ca),吡啶基(3cj,3ck)和嘧啶基(3cl)都是耐受的。两个螺环氮杂环丁烷(3cm, 3cn)的成功胺化进一步证明了该方法的相容性。此外,5至7元环状和桥环胺,包括吡咯烷(3co-3ct)、吗啉(3cu)、四氢异喹啉(3cv)、氮杂环庚烷(3cw)和二氮杂双环[2.2.1]庚烷(3cx)也是适合的,以高达91%的收率提供相应的产品。无环仲胺以中等收率产生所需的产物(3cy-3de)。然而,由于空间位阻,叔丁胺(3bj)和二异丙胺(3df)未能进行所需的N-芳基化。
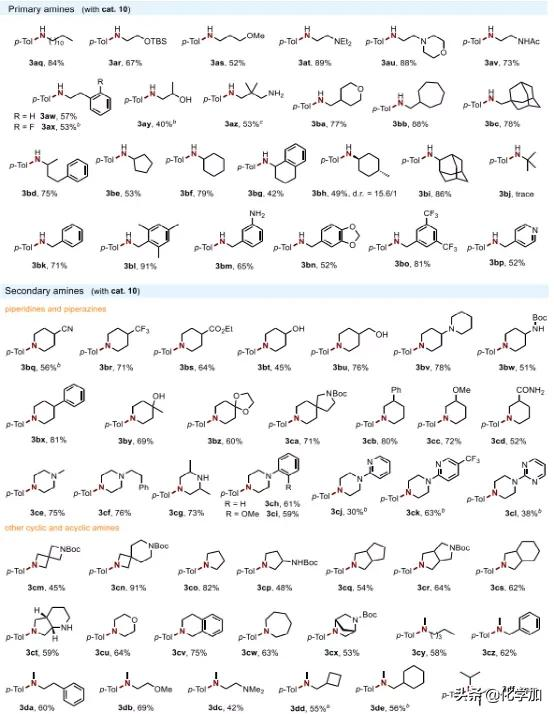
图3. 胺的底物范围(图片来源:J. Am. Chem. Soc.)
该反应的高度相容性鼓励作者研究其在后期功能化中的实用性(图4a)。Paracetamol(扑热息痛)是一种常见的镇痛解热药,hordenine(大麦牙碱)可缓解支气管炎症状,它们都是合适的底物,分别提供4a和4b。双酚A的胺化提供了70%的单胺和二胺产物(4c)。缩酮-雌酮和17-庚酸雌二醇也与反应条件相容,分别得到4d和4e。此外,该方法成功地用于一系列药物的 N-芳基化,包括伯胺美西律(4f)和脱氢枞胺(4g)、无环仲胺氟西汀(4h)、哌啶帕罗西汀(4i)和哌嗪阿莫沙平(4j)。
图4. 后期官能团化及机理研究(图片来源:J. Am. Chem. Soc.)
为了深入了解机理,作者监测了4-甲基苯酚(1a)和哌啶(2a)之间的催化胺化反应,并观察到 2a的逐渐消失和苯胺3a的形成;没有检测到任何亚胺、环己酮或环己烯-1-酮(图4b)。此外,1a与α-氘代十二胺(2aq)胺化生成苯胺3aq(95%氘掺入),在12小时内氘掺入没有明显衰减;然而,当反应进行24-36小时(图4c)时,观察到衰减。值得注意的是,没有检测到芳族H/D交换,表明这种胺化的氢转移机制不太可能。当η5-phenoxo络合物Rh-1由相应的Cp*Rh η6-苯酚络合物在碱存在下通过易于去质子化形成的,经受胺化条件(图4d),以55%的产率获得游离苯胺3af和3,5-二甲基苯酚(1af)。此外,η6-苯胺络合物Rh-2和1af之间的芳烃交换反应以几乎定量的产率产生η5-苯氧络合物Rh-1和游离苯胺。这些结果支持图1c中描述的π配位激活机理。
总结:西湖大学石航教授课题组开发了一种在没有外源活化试剂的情况下通过铑催化的酚和胺之间的脱水缩合反应简明合成苯胺的新策略。鉴于该方法在苯酚和胺方面的广泛范围及其广泛的官能团耐受性,将具有广泛的合成效用。