

 在线客服
在线客服
 快速发布
快速发布
 我的店铺
我的店铺
 我的化学加
我的化学加
 我的消息
我的消息
 我要充值
我要充值


 回到顶部
回到顶部



买产品

克难题

技术供需

求职招聘

发定制

项目整合

园区推荐

行业资讯

化学加智库

商家
买产品
克难题
技术供需
求职招聘
发定制
项目整合
园区推荐
行业资讯
化学加智库
商家
相比于传统的无机铁电材料,有机铁电材料拥有高柔韧性、抗崩损性和溶液可加工性的优点,因而是国内外一个重要的研究领域。在目前已开发的有机铁电材料中,无金属钙钛矿铁电材料已达到与传统无机铁电材料相当的铁电极化率,但它们存在一个严重缺陷:其矫顽力普遍仅为~10kV/cm。这导致在器件设计中,这些有机铁电材料的厚度要达到约1μm才能保证电压转变区间为1~2V,因而阻碍了器件的微型化。一般而言,理想的矫顽力需在~100kV/cm量级,但目前仍缺乏有效手段来实现该目标。
近日,北京大学深圳研究生院新材料学院潘锋教授团队与新加坡国立大学罗健平教授团队在《自然•通讯》(Nature Communications,2022,13:794)杂志上发表了题为“Tailoring the coercive field in ferroelectric metal-free perovskites by hydrogen bonding”的研究论文。该研究提出了一种基于氢键作用的矫顽力调控策略,可以使无金属钙钛矿铁电材料的矫顽力从~10kV/cm量级提升至~100kV/cm,为有机铁电器件微型化和集成化设计提供了一条重要途径。

有机铁电材料的分子结构和氢键相互作用
研究团队设计并合成了两种无金属钙钛矿铁电材料MDABCO-NH4-(PF6)3 (MDABCO=N-methyl-N'-diazabicyclo[2.2.2]octonium)和MDABCO-NH4-I3,分别称为MNP3和MNI3。其中,有机分子MDABCO的转动可以实现有序-无序排列的转变,从而带来材料的铁电-顺电相变。测量结果表明,MNP3中的N–H…F氢键距离远低于MNI3中的N–H…I氢键,该氢键导致MDABCO的偏移量高于MNI3。这种氢键作用使材料的矫顽力从MNI3的12kV/cm提升至MNP3的110kV/cm,实现了有机铁电材料矫顽力调控的目标。
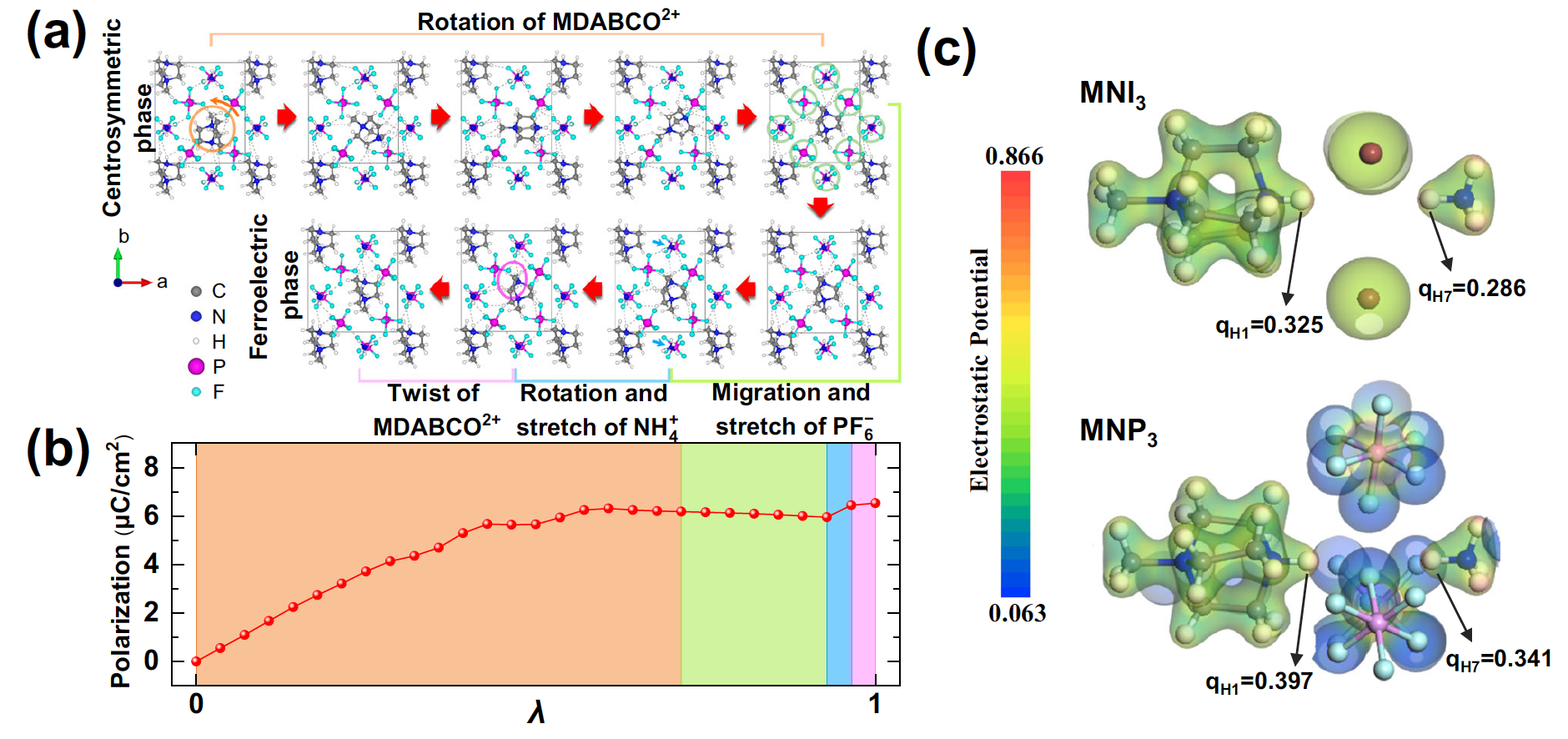
基于氢键有机铁电材料的理论计算和相关机理
研究团队进一步通过同位素实验和第一性原理计算探讨氢键作用影响矫顽力的内在机理。他们将MDABCO分子中N-H上的H替换为氘(D),发现MNP3的矫顽力可从110kV/cm提升至138kV/cm。同时,晶胞沿[111]方向有明显拉伸,说明氢键对MDABCO分子的作用力显著。通过对氘化前后的MNP3进行铁电相变过程的测量,发现居里温度在氘化后有所上升,说明了氢键对MDABCO分子运动的限制。核磁共振波谱测量证实MDABCO与PF6−之间的氢键可以直接影响两者的结构变化和运动。压电响应力显微镜观测到四个铁电畴,其在正负电压切换过程中的180度畴翻转和蝴蝶曲线说明MNP3具有良好的铁电性。第一性原理计算结果证实PF6−相比于I−对MDABCO上的电子有更强的吸引力。Berrry相计算获得了与实验测量值吻合的理论值,且揭示了MDABCO在转动过程中会伴随着N–H…F氢键的断裂和形成。这些结果均表明,当材料内有机分子MDABCO所受到的氢键作用被有效增强后,该分子在铁电转变过程中的转动将会明显受阻,从而使材料的矫顽力提升一个数量级。
新加坡国立大学的Hwa Seob Choi博士和北京大学深圳研究生院新材料学院的李舜宁副研究员为该论文的共同第一作者,潘锋和罗健平为通讯作者。该研究得到了广东省重点实验室及软科学研究计划项目的支持。
参考资料:https://news.pku.edu.cn/xsky/6ec58ab7346c4b7883b8bacd6c00bbc6.htm
声明:化学加刊发或者转载此文只是出于传递、分享更多信息之目的,并不意味认同其观点或证实其描述。若有来源标注错误或侵犯了您的合法权益,请作者持权属证明与本网联系,我们将及时更正、删除,谢谢。 电话:18676881059,邮箱:gongjian@huaxuejia.cn
