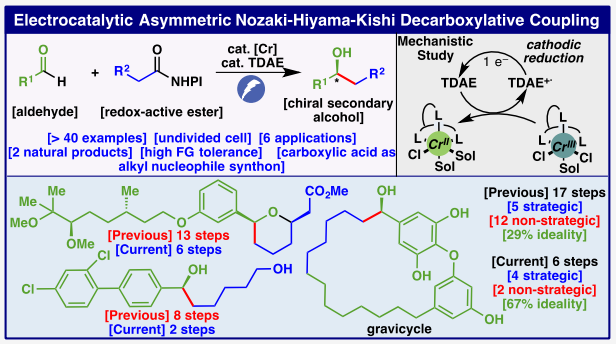
40多年来,手性仲醇的合成一直是化学家们研究的热点(Figure 1A)。合成芳基-烷基取代仲醇的两种主要途径是对醛进行亲核加成和对酮进行不对称还原。Nozaki-Hiyama-Kishi反应于1977年首次发现,其使用化学计量的Cr和催化量的Ni,通过烯基卤化物与醛的交叉偶联来实现烯丙醇产物的合成。然而,该反应却很少用于烷基卤化物。近年来,尽管化学家们发展了一系列烷基亲核试剂替代物,如烷基碘化物,羧酸(通过氧化还原活性酯(RAEs)),烯烃,甚至非活化C-H键(Figure 1B)。然而到目前为止,还没有以高度对映选择性实现该转化的报道。最近,美国加州理工学院Sarah E. Reisman课题组和美国斯克里普斯研究所Phil S. Baran课联合报道了首例对映选择性烷基-Nozaki-Hiyama-Kishi偶联反应。其使用Cr-电催化的脱羧策略,在温和的电催化还原条件下以良好的产率和对映选择性实现了一系列手性仲醇的合成(Figure 1C)。下载化学加APP到你手机,收获更多商业合作机会。首先,作者对反应条件进行了优化(Table 1)。当使用醛4(0.8 mmol),氧化还原活性酯5(2.0 equiv),在CrCl2(30 mol%),L7(33 mol%),Proton Sponge (33 mol%),TESCl(2.0 equiv),TDAE(0.4 equiv),TBAClO4(0.1 M),(+)Al/(-)Ni foam,MeCN (c = 0.5 M),2.5 mA,室温反应24小时后用TBAF后处理,可以以58%的核磁产率,90% ee得到相应的手性仲醇产物6。 在得到了最优反应条件后,作者对此转化的底物范围进行了探索(Table 2)。实验结果表明,一系列不同取代的氧化还原活性酯与醛均具有良好的兼容性,以32-71%的产率,80-91%ee得到相应的手性仲醇产物7-47。值得注意的是,此转化对一系列生物活性分子骨架,如linoleic acid, chlorambucil, elaidic acid, naproxen等均具有良好的兼容性。遗憾的是,此转化对硝基、苯腈以及吡啶基不能兼容(49-51)。 利用作者所发展的电催化不对称NHK脱羧偶联策略,可以大大简化分子的合成路线。这主要是由于此策略采用了基于自由基的逆合成策略,这与传统用于合成仲醇的传统2e-策略有所不同。例如,之前的合成方法需要经历包括官能团转换等五步来实现炔54的合成,而利用此策略可以从53开始仅用两步即可实现54的制备(Figure 2A)。天然产物全合成中的二醇中间体58,传统方法制备消旋的产物需要经历六步,而利用此方法仅需三步即可以良好的对映选择性实现手性产物58的合成(Figure 2B)。对于药物相关的二醇63,之前的策略需要八步,而现有策略仅需两步(Figure 2C)。值得注意的是,使用简单易得的RAE 64,仅通过两步即可实现horsfieldone A的首次全合成(Figure 2D)。 此外,利用此策略还可以实现更复杂分子的合成(Figure 3)。例如,herboxidiene类似物70,之前报道的方法需要经历十三步合成。而利用醛68作为起始原料,通过所发展的e-NHK偶联(35%,94:6 dr)以及随后的环化(44%),仅需六步即可实现70的合成(Figure 3A)。而之前所报道的gravicycle (78)的十七步合成也可以利用此策略,仅使用简单的芳基碘74作为起始原料,通过与RAE 75的电催化DCC-芳基化(51%)、与76的Ullman偶联、与RAE 77的e-NHK(46%,82%ee)、RCM(71%)以及脱保护(92%)六步得到目标产物78(Figure 3B)。 最后,作者对此转化的反应机理进行了探索(Figure 4),并得出如下结论:1)无论化学计量反应还是催化反应条件均涉及到假定的烷基铬物种的形成,并且该中间体的形成不需要TDAE。因此,在电催化体系中,TDAE介导了L7·CrIII的还原。2)CV实验表明,配体增加了络合物的还原电位或增加了过电位(overpotential),并进一步证实了TDAE的作用是介导L7·CrIII络合物的还原。 Sarah E. Reisman和Phil S. Baran课题组联合报道了首例Cr-电共催化的对映选择性烷基-Nozaki-Hiyama-Kishi (NHK)偶联反应。该反应利用简单的伯烷基底物与醛的加成,高对映选择性的实现一系列手性仲醇的合成。此不对称烷基e-NHK反应实现的关键是通过使用TDAE作为关键的还原介质。CV研究和化学计量实验表明,TDAE的作用是介导L7·CrIII络合物的还原,而在之前的非不对称烷基e-NHK中,这一过程是决速步骤。而这对于不对称反应特别有利,由于在不对称反应中,手性配体在电极处实现缓慢地的催化剂的还原。因此使用催化量TDAE介导对于避免烷基自由基和TDAE+•之间的竞争HAT过程至关重要。此外,该策略可以有效减少多种药物相关结构和天然产物的合成步数,进一步证明了此转化的实用性。 文献详情:
Yang Gao, Baiyang Jiang, Nathan C. Friede, Arianne C. Hunter, Dylan G. Boucher, Shelley D. Minteer, Matthew S. Sigman, Sarah E. Reisman*, Phil S. Baran*. Electrocatalytic Asymmetric Nozaki–Hiyama–Kishi Decarboxylative Coupling: Scope, Applications, and Mechanism. J. Am. Chem. Soc., 2024, https://doi.org/10.1021/jacs.3c13442.


 在线客服
在线客服
 快速发布
快速发布
 我的店铺
我的店铺
 我的化学加
我的化学加
 我的消息
我的消息
 我要充值
我要充值


 回到顶部
回到顶部


















