

 在线客服
在线客服
 快速发布
快速发布
 我的店铺
我的店铺
 我的化学加
我的化学加
 我的消息
我的消息
 我要充值
我要充值


 回到顶部
回到顶部



买产品

克难题

技术供需

求职招聘

发定制

项目整合

园区推荐

行业资讯

化学加智库

商家
买产品
克难题
技术供需
求职招聘
发定制
项目整合
园区推荐
行业资讯
化学加智库
商家
“重磅炸弹”新药合成与工艺绿色化
Blockbuster Drugs Synthesis and Greening Pharmaceutical Process
张 霁1,2,张英俊1,2,聂 飚1
(1. 广东东阳光集团药业研究院,广东东莞 523871;2. 抗感染新药研发国家重点实验室,广东东阳光药业,广东东莞 523871)
摘要:通过评述CDK4/6 抑制剂帕博昔布、丙肝NS3/4A 蛋白酶抑制剂司美匹韦和NS5A 抑制剂Elbasvir 等“重磅炸弹”新药合成及工艺过程,对其中工艺研究的重点难点和瓶颈反应进行全面分析,揭示关键反应的优化以及分离纯化节点的选择实施,是整个工艺绿色化的重点。同时对手性相转移催化技术、酶催化方法和SFC 分离技术等绿色手段在合成工艺中的应用进行了案例解析。总结出一些基本的工艺特点和规律, 最后展望了工艺绿色化的一些新前沿、新亮点。
关键词:“重磅炸弹”药物;绿色工艺;工艺优化;药物合成

1 前言
“重磅炸弹”新药一般是指年销售额达10 亿美元以上的药物[ 1],如辉瑞的MG-CoA 还原酶抑制剂立普妥(Lipitor)、艾伯维的阿达木单抗(Humira)以及默沙东的二肽基肽酶-4(DPP-4) 抑制剂西格列汀( sitagliptin) 等。由于这类药物的作用机制新颖、疗效优异,上市后迅速被医生和患者接受使用,因此获得的社会及经济效益都非常可观。与此同时,这些重磅药物均由世界一流的制药企业研发和生产,其开发的合成工艺方法也是非常值得学习和借鉴的。例如西格列汀的化学工艺就曾2 次获得美国绿色化学的最高奖(Presidential Green ChemistryChallenge Award)[2—4]。本文就新近出现的“重磅炸弹”药物的合成路线及工艺进行分析,探讨新药合成工艺开发的过程和绿色化的途径[5]。
2 帕博昔布(palbociclib,17) 的合成与工艺绿色化
17 是全球首个上市的CDK4/6 抑制剂[6]。基于Ⅱ期临床无进展生存期(progression free survival,PFS) 的优秀疗效数据,2015 年被FDA 加速批准,
用于治疗HR+/HER2- 晚期乳腺癌,第一年即实现了7.23 亿美元的销售额,2016 年上半年全球销售额达到9.42 亿美元,预计全年销售额可达23 亿美元,已成为名副其实的“重磅炸弹”新药。该药物发现的初始路线( 图1) 是从4- 氯-2- 甲硫基嘧啶-5- 甲酸乙酯(1) 出发,经过7 步反应构建溴代吡啶并嘧啶酮母核中间体10,然后经过特别的氧化剂11 处理,得到了关键中间体亚砜12,随后的亲核取代反应连接氨基吡啶13,接着在Pd(PPh3) 4催化下,利用Still 偶联反应制备了烯醇醚16,经盐酸酸性水解除去保护基,获得了17 的盐酸盐。该路线共涉及11 步反应,总收率仅4.8% [7]。
经过考查和前瞻性分析,不难发现该路线有以下不足和缺陷:①该路线需使用易燃易爆的氢化铝锂(LiAlH4),以及纯度和品质无法确定、转移困难和有加料风险的氢化钠(NaH);②中间体多次不合理的氧化态改变(LiAlH4 还原-MnO2 氧化- 格氏试剂加成-NMO/TPAP 氧化- 硫醚的特殊氧化成亚砜等);③采用非绿色的Horner-Wadsworth-Emmons反应用来构建吡啶并嘧啶酮杂环母核;④ Still 偶联反应使用有毒的锡试剂,残留锡不易彻底消除,而且会产生极性和溶解度与产物相似的杂质,难以分离纯化;⑤需要柱色谱分离纯化中间体7、12 和16等,难以实现大规模工业化生产;⑥值得一提的是,罕见的氧化剂即希夫碱环氧化物11 制备极其困难,稳定性极差、易分解,纯化工艺及纯度都不好控制,储存也是难题,更麻烦的是,用该试剂制备的亚砜反应活性不高,随后的亲核取代反应产率很低(38% ),严格来说该反应是整个过程的第一个瓶颈反应(bottleneck reaction) [8]。

辉瑞的工艺研发者对这个瓶颈反应进行了全面攻关,发现当X=MeSO2 时,亲核取代反应有所改善( 产率增至50% ),但优化反应条件后收率仍不理想。考虑到制备化合物10a 和10b 步骤多(10 ~11 步),故决定迅速开发以X=Cl 为中间体的关键反应( 图2),因为该中间体22 仅需4 步就能方便有效地从5- 溴-2,4- 二氯嘧啶(18) 制备( 图3):①温和条件下的选择性胺化;②区域选择性Heck 反应;③ NBS 溴代反应得到溴代吡啶并嘧啶酮母核中间体[9]。

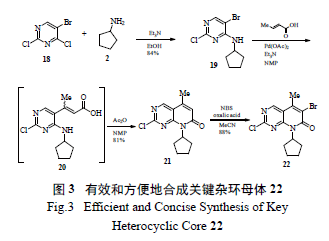
随着化合物22 的成功制备,氨基吡啶和化合物22 的高度区域选择性SNAr 偶联便成了关键反应(key reaction)。辉瑞工艺研究人员通过系统的研究发现,许多文献报道的钯催化C-N 偶联反应在此处的应用并不理想,通过筛选不同的钯盐[ 例如Pd(OAc)2、Pd2(dba)3、POPd]、膦配体(BINAP、DavePhos、JohnPhos)、碱(NaOt-Bu、K3PO4、Cs2CO3)以及溶剂( 甲苯、DME), 最好的收率也仅为40%。考虑到钯催化剂成本较高,且实验结果难以令人满意,故研究人员认为该法不切实际,难以用于放大生产。
相对于亚砜10a 和砜10b,氯代的吡啶并嘧啶-7- 酮22 的反应活性很低,且氨基吡啶是一个弱的双向的亲核试剂(ambident nucleophile),在中性条件下即便是在回流的甲苯里,也得不到预计的产物14。相反,在弱碱条件下,却能得到少量的区域异构体23( 图4),这也说眀氨基吡啶杂环上的氮亲核活性更高。显然如何将区域异构体23 转变为理想产物14 也是必须面对和解决的一个难题。
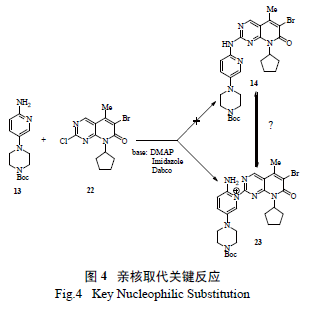
随后的试验表明,区域异构体23 在强碱性条件下,通过调节pH 值或淬灭反应,可转化为理想产物14。欣喜的是,在线红外(ReactIR) 光谱跟踪检测发现,在强碱LiHMDS 作用下,反应物13 能脱质子形成氨基吡啶负离子,能有效制备14,当加入过量的碱(2.5 倍摩尔量) 和13(2.2 倍摩尔量) 时,反应转化率能从28%提高至85%。美中不足的是,在过量的强碱条件下,14 不稳定,会产生二聚体副产物24( 1,6- 加成产物),给分离纯化带来极大的困扰( 图5)。如降低碱的用量( 小于2 倍摩尔量),则转化率低于50%。显然,从原子经济学和产物的转化率、分离纯化等多方面考虑,使用LiHMDS 作碱仍不是最佳选择。
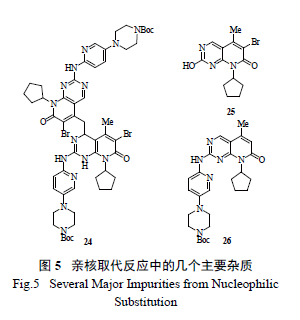
初始的反应动力学研究认为该亲核取代反应是一个二级反应( 图6),这能够解释为什么中间体形成后,需要另一分子的锂化氨基吡啶负离子作进攻试剂。但是随后的反应转化率数据和动力学模型DynoChem 计算发现,二级反应的机理和数据不能与实验结果很好匹配,相反,反应数据却能和一级反应很好吻合,故认为该反应是经过四元环的过渡态,即经过[1,3] 重排迁移完成的( 图7)。
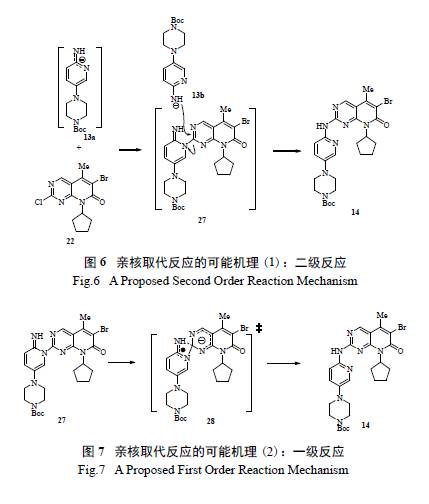
HOMO 的计算也表明,在中性条件下,化合物13a 中吡啶环上的氮和环上的氨基有大致相同的亲核活性,许多情况下,产生区域异构体27 显然是动力学上有利生成的。由于环外的氮有较高的电子云密度,且不易离域化,故通过使用其他的强碱试剂,极有可能形成脱质子的氨基负离子。初步筛选强碱,如NaOtBu, KOtBu, NaOt-Am 可得到约30%~ 40%的理想产物14,但不可避免地带来水解副产物25。若严格干燥各种试剂并小心处理反应,可以有效控制杂质生成,收率能提高到87%,但在大规模的放大生产中,完全严格地除水是不现实的。由此,辉瑞研发团队决定使用氯化异丙基镁作为碱参与亲核取代反应,结果反应进行得非常干净彻底,且未生成二聚体副产物24。当13 和格氏试剂摩尔比1.1 ∶ 2 时,收率81%;优化后摩尔比调整为1.3 ∶ 2.2 时,收率提高到94%,但有少量脱溴副产物形成。他们通过在线红外和离线分析发现反应机理存在2 种可能的途径:(1) 直接亲核取代;( 2) 反应机理和前述相同,但可能中间体23 转化为产物14 太快,无法用在线红外跟踪检测。鉴于用氯化异丙基镁作碱在反应过程中会产生异丙烷气图6 亲核取代反应的可能机理(1):二级反应Fig.6 A Proposed Second Order Reaction Mechanism图7 亲核取代反应的可能机理(2):一级反应Fig.7 A Proposed First Order Reaction Mechanism体,在最后的优化工艺中,改用氯化环己基镁,解决了产生气体所带来的安全隐患,已成功应用于实际生产。
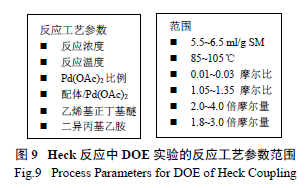
最后,利用钯催化的Heck 反应引入烯醇醚成为整个工艺的另一个关键反应。研发人员通过DOE实验[10],系统全面地优化了实验参数,合理化了可操作的实验空间,进一步抑制和控制了杂质( 图8) 含量,这包括催化剂的选择、50 个不同的配体和5 个不同碱的筛选组合,一共进行了250 个反应( 图9),分别找出了脱溴杂质26 产生的敏感因素[Pd(OAc) 2 与温度关系图];乙烯基杂质27 和区域异构体28 产生的敏感因素[DPEphos 配体与Pd(OAc)2 的关系图],真正将反应的控制从定性的范围提高至一个定量的高度。优选后的反应将26、乙烯支链杂质29 和区域异构体30 控制在合理范围,同时使这个关键反应能够在一个极为合理的参数空间三维进行操作,而不是一个局部二维的优化。
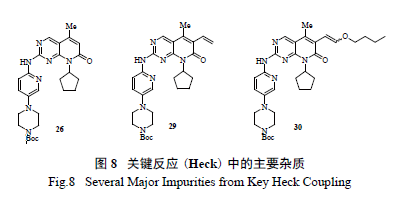
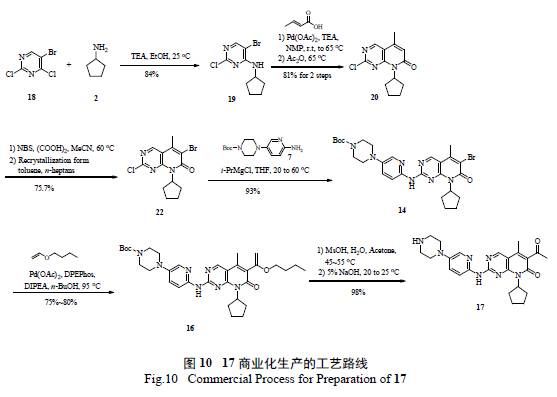
综上所述,不难发现辉瑞工艺研发人员通过对17 药物发现路线的认真评估和研究,通过引入更加合理易得的原料,首先迅速克服了瓶颈反应的难题,随后将2 个关键反应( 亲核取代和Heck 偶联) 进行合理分析、优化和动力学研究,找到了合理操作及控制的空间,避免了杂质的大量形成,简化了反应的后续分离纯化,解决了一系列安全隐患。将反应从原来的11 步简化到可商业化生产的7步( 图10),总收率也从初始的4.8%改善到目前的37.7% [11]。总而言之,整个工艺的绿色化表现在:①合成策略和关键中间体的正确选择;②非绿色、不安全反应的弃用;③现代PAT 技术和动力学研究辅助工艺优化;④ DOE 实验优化反应;⑤ Heck 反应置换Still 偶联反应;⑥避免不合理的氧化还原反应过程。
3 丙肝NS3/4A 蛋白酶抑制剂司美匹韦的合成及工艺绿色化
2014 年上市的吉利德史诗级“ 重磅炸弹”NS5B 抑制剂索非布韦( sofosbuvir) 以优异的治疗效果、广谱的抗病毒活性、高耐药屏障以及极低的交叉耐药可能性,迅速占领了市场,2015 年全年销售53 亿美元,sofosbuvir 和ledipasvir 的复方Harvoni 全年达139 亿美元,创造了销售奇迹[ 12]。杨森制药开发的司美匹韦( simeprevir) 是第二代NS3/4A 抑制剂,它的出现对NS3/4A 抑制剂的开发是革命性的,不仅表现出对NS3/4 蛋白酶极高的抑制活性[13],而且临床上与干扰素/ 利巴韦林合用的三联疗法治愈率极高,12 周的持续病毒响应时间高达88.6%,相对之前的疗法有了极大提高,同时治疗周期也缩短到12 周。2014 年也创下了年销售23亿美元的佳绩。其他一些丙肝NS3/4A 抑制剂的结构见图11。
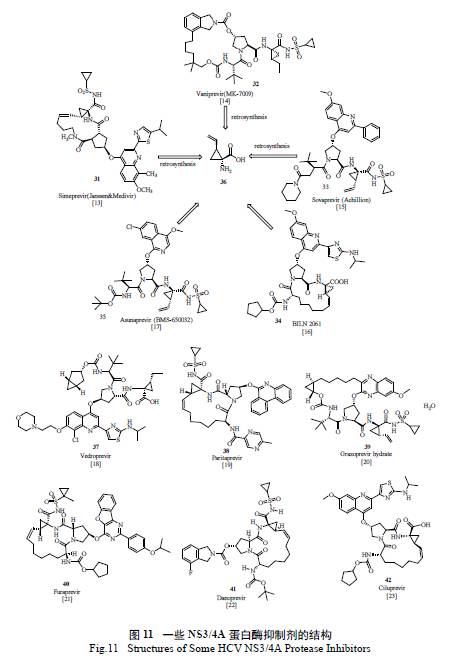
从图11 来看,这些抑制剂都含有一个共同的手性小分子,即保护的(1R,2S) -1- 氨基-2- 乙烯基环丙基羧酸,Boehringer-Ingelheim(BI) 的化学家首先发展了一种以价廉易得的甘氨酸酯盐酸盐为起始原料的简约方法( 图12):选择1,4- 二溴代-2- 丁烯为另一原料,利用串联的SN2-SN2' 双烷基化过程,一步到位极为便利地制备了外消旋的氨基酸。值得一提的是,作为关键反应,反应条件的选择和优化对控制主要副产物48 的形成至关重要。通过数十个反应条件的仔细筛选,将理想产物45 与48 的比例从极不理想的1.2 ∶ 1 优化至>40 ∶ 1,易于分离纯化,也为叠缩工艺(Telescoping Process) 的实施打下了坚实的基础[24]。

从工艺绿色化的角度来分析整个流程,不难发现该工艺对每个基元反应的研究和优化都很透彻,虽然前面数个反应的产物、副产物和原料都是液体,但通过反应的高产率、高效辅以理想的中控,避免了3 个液体中间体的分离纯化,实现了叠缩工艺的完美结合,成功完成了从公斤级实验到中试的跨越。而利用酶催化选择性在温和条件下拆分外消旋氨基酸,也是绿色工艺的很好体现。
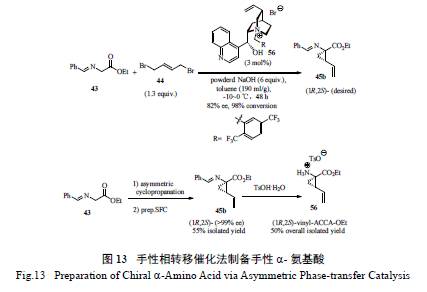
在BI 的工作基础上,Merck 的工艺化学家巧妙使用了无需过渡金属催化的手性相转移催化绿色工艺,用以甘氨酸希夫碱和二溴丁烯为原料的合成方法[25],一步到位构筑了含有环丙烷基和乙烯基的手性α- 氨基酸酯( 图13)。值得一提的是,通过对反应条件的筛选和优化( 手性相转移催化剂的用量、碱的选择、水的用量控制以及反应时间和温度等),取得了较为理想的效果,并结合制备型手性柱的超临界流体色谱(SFC) 分离(SFC 是一种提高分离效率的绿色环保关键技术),以55%的分离产率、>99% ee 的光学纯度制备了化合物56。
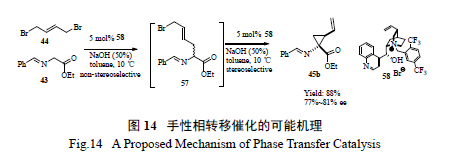
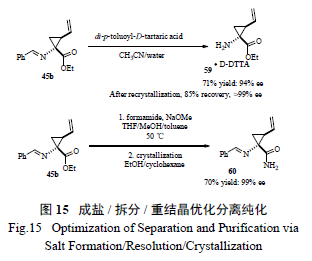
百时美施贵宝工艺小组仔细研究了在手性相转移催化剂催化下进行不对称环丙基化反应的机理[26],发现第一步烷基化反应并不是立体选择的,而随后的关环反应形成环丙烷则是立体选择的( 图14)。该小组分析了催化剂的降解途径,通过对化学过程的透彻研究,采用切实可行的拆分- 重结晶和酯的直接酰胺化- 重结晶过程,开发出一条简洁有效、不需要柱色谱分离的工艺( 图15),有效制备了关键手性环丙烷α- 氨基酸中间体。值得强调的是,最开始反应得到的中间体为液态,但工艺研发人员一般偏爱处理固体物质( 因为其分离纯化手段多,且易于放大操作),因此利用成盐进行拆分、打浆提高纯度以及随后的酰胺化得到固体,不失为工艺绿化的良策。
综合上述3 家制药公司对该反应系统的深入研究,不难发现其对关键反应的重视,同时也体会到反应及工艺绿色化是一个逐步完善的过程。绿色化方法如酶催化(BI)、SFC 分离技术(Merck),叠缩技术(BI) 和手性相转移催化(Merck, BMS) 以及成盐打浆(BMS, Merck, BI) 等实用技术都能得到很好地利用[27—29]。
4 烯烃复分解反应在合成大环内酯/ 内酰胺中的应用和工艺绿色化
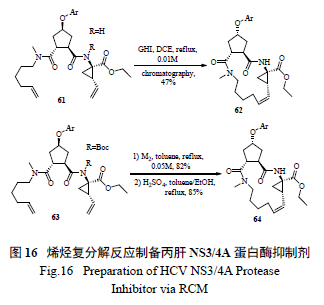
很多大环内酯和大环内酰胺药物( 如克拉霉素和阿奇霉素) 都是重要的抗感染药物,与普通百姓的健康息息相关。过去这类药物的合成有的采用传统的酯化和酰胺化,不仅产率、纯度低,而且分离纯化非常困难。近年来,烯烃复分解反应得到了广泛利用,并逐渐成为常用的绿色化工艺手段和方法( 图16)[30—31]。
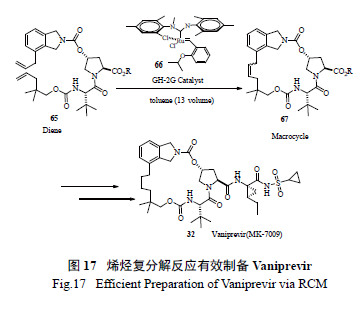
使用烯烃复分解反应最为成功的案例当属Merck 制药的工艺研究小组将其用于HCV 新药研发,该小组釆用同时缓慢滴加Ru 催化剂和二烯底物的方法,极为出色地生产了药物Vaniprevir(MK7009) [32]。该策略的亮点为:一是催化剂的用量很少(0.2 mol% ),二是底物浓度高(0.13 mol/L),不需要高度稀释;三是极为优秀的高产率(91% )。这也是催化手段绿色工艺化学的极佳案例( 图17)。
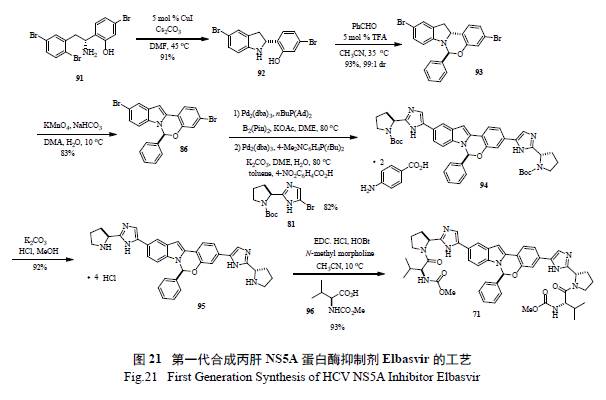
5 丙肝NS5A 蛋白酶抑制剂Elbasvir 关键反应的绿色化
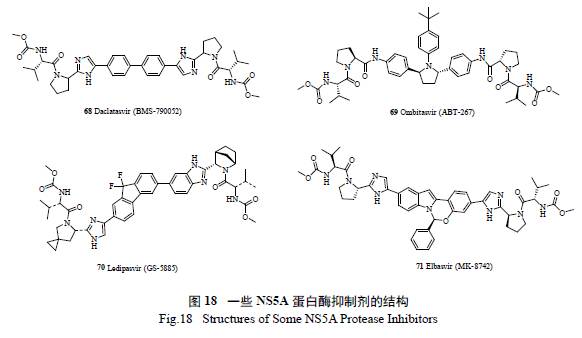
ZEPATIER(Elbasvir / Grazoprevir) 是一款口服丙肝药物组合[ 其中Elbasvir(71) 是一种NS5A抑制剂[33],Grazoprevir 是一种NS3/4A 蛋白酶抑制剂[34] ] 可有效抑制丙肝病毒的复制,2016 年1 月28 日被FDA 批准用于治疗慢性丙型肝炎(HCV)基因1 型和4 型感染。该联合药物不需要联合使用干扰素,避免了干扰素治疗可能产生的严重不良反应。预计2017 年ZEPATIER 的销售额有望达到15 亿美元。图18 是FDA 批准上市的NS5A 蛋白酶抑制剂的结构。
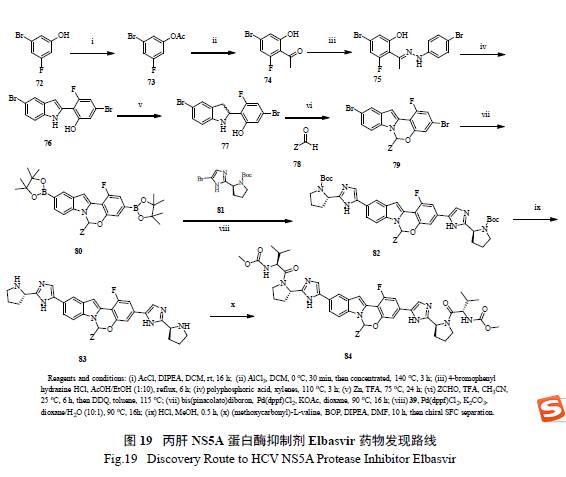
71 的发现路线如图所示( 图19),从简单的卤代苯酚出发,经过10 步反应,制得71 及其类似物,该法能够方便快捷地合成一系列化合物,有效进行SAR 构效关系研究[35]。仔细考查71 结构,发现其中心结构(core) 两边所连接的基团是一样的,如果中间体选择合适,分子的局部对称性对简化合成是有帮助的。值得强调的是整个合成的重点是如何有效地制备光学化活性半缩醛胺(hemiaminal),这个结构在药物分子中是极为罕见的。
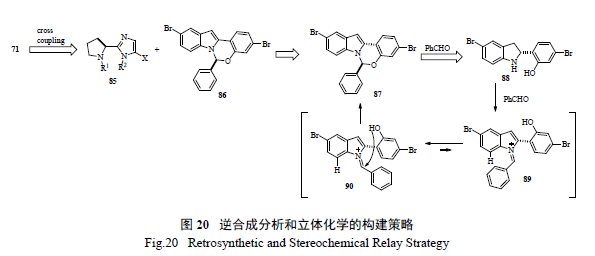
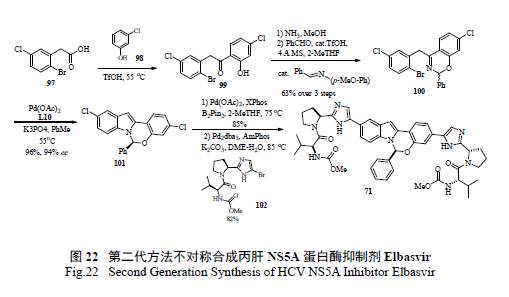
Merck 工艺研究人员通过逆合成分析( 图20)和对反应机理的深度理解,认为最重要和最富有挑战的是如何有效的构造半缩醛胺的手性中心。如图21 所示,光学活性的二氢吲哚的制备,以及随后的和醛加成消除制备活泼亚胺正离子都是至关重要的。这样从光学活性的胺出发,经过9 步反应,有效制备了71[36]。美中不足的是,手性胺的制备是通过不对称氢转移来实现的,而通过氧化反应将二氢吲哚转变为吲哚显然有违于绿色化学的原理。在合成工艺研究中,对关键反应的攻关和突破对整个工艺的绿色化举足轻重。Merck 工艺化学家们创造性地发展了一个新颖独特的化学反应,高效地制备了光学活性的半缩醛胺[37]。如图22 所示,其优秀之处表现在:(1) 釆用价廉易得的起始原料;( 2) 方便有效地制备外消旋苯并噁嗪,并通过其开环互变异构和手性双膦单氧化物存在下的钯催化Buchwald-Hartwig C-N 键的偶联反应来控制半缩醛胺手性中心的形成;( 3) 通过对反应条件的广泛筛选和优化,该关键反应的结果非常理想,ee 值高达94%,产率96%;(4) 避免了不必要的二氢吲哚和吲哚之间的转换;( 5) 起始原料均为氯代苯而不是溴代苯,从原子经济的原则来判断后续的偶联反应,也是更趋环保绿色的途径。简而言之,这是一条绿色化的工艺合成路线,必将成为合成与工艺绿色化的杰出案例。
6 结语与展望
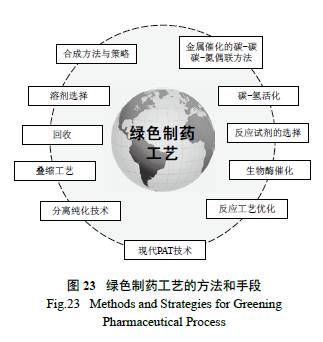
制药工艺的绿色化是一个复杂、逐渐优化的系统工程( 图23),是将一系列反应有效地“串联”起来,从简单易得的原料来制备复杂药物分子的过程。涉及合成化学单元反应的有效连接,是一个“承上启下”的科学活动[ 38]。因此,它不仅要考虑合成方法与策略、绿色的手段( 金属催化的偶联反应、C-H 活化、生物酶催化),而且追求实现完美工艺的技巧( 如工艺参数的优化、叠缩工艺的使用等)、分离的便利有效( 成盐方式或打浆拆分) 以及溶剂的选择与回收等。应该说,追求制药工艺的完美和绿色化是业界同行的共识[39—40]。
最后,我们期待本文中分析总结的一些重磅新药的绿色工艺的技巧和方法,能为业界同行的实际工作提供有益的借鉴与思考。同时希望国内制药同仁不断向跨国制药的一流水平看齐,提高技术能力,开发出更加环保便捷、经济实用的药物绿色合成新方法、新工艺,为造福人类、创造更加文明的生态环境做出应有的贡献。
致谢:本文是根据张霁博士在深圳南方科技大学举办的2016 中国制药化学反应及工艺高峰论坛的报告整理而成。感谢张绪穆教授和周伟澄研究员给予的支持和建议
基金项目:广东省引进创新创业团队计划(No.201301Y0105381261)
作者简介:张 霁(1962—),男,博士,药物研发/工艺研发首席科学家,曾任职于跨国药业雅培(Abbott)、辉瑞(Pfizer)和百时美施贵宝(Bristol-Myers-Squibb),从事新药研发和工艺研究。
声明:化学加刊发或者转载此文只是出于传递、分享更多信息之目的,并不意味认同其观点或证实其描述。若有来源标注错误或侵犯了您的合法权益,请作者持权属证明与本网联系,我们将及时更正、删除,谢谢。 电话:18676881059,邮箱:gongjian@huaxuejia.cn
