

 在线客服
在线客服
 快速发布
快速发布
 我的店铺
我的店铺
 我的化学加
我的化学加
 我的消息
我的消息
 我要充值
我要充值


 回到顶部
回到顶部



买产品

克难题

技术供需

求职招聘

发定制

项目整合

园区推荐

行业资讯

化学加智库

商家
买产品
克难题
技术供需
求职招聘
发定制
项目整合
园区推荐
行业资讯
化学加智库
商家
电催化分解水(HER)是获取氢气能源最重要的手段之一,由于该技术的最终产物为高能量密度的氢气与无污染的氧气,使得以水分子为载体的能源储存过程满足可持续绿色能源的要求。立方型过渡金属氮化钼因具有较好的电化学稳定性、水吸附特性,是理想的HER催化材料,然而作为金属-非金属合金化合物,其本征间隙空位的存在极大地减小了金属d带中心与费米能级的距离,使得材料对于氢物种的吸附过强,限制了其HER活性。
基于此,作者首先利用DFT理论计算预测了一种无本征间隙空位的化学计量型碳氮化钼晶体结构(图1)。电荷局域密度分布(ELF)、态密度分布(PDOS)、Bader电荷和差分电荷等计算结果表明:随着晶格碳原子对氮化钼材料本征间隙空位的逐步取代,钼原子未配位的价电子将向碳原子发生转移,自由电子趋向于局域化,进而推动C 2p和 Mo 4d轨道的杂化耦合,降低材料的d带中心,减弱其对氢物种的吸附作用;与此同时,钼原子失去更多的价电子,氧化态上升,水分子的吸附解离过程得到优化;更重要的是晶格碳原子周围更低的轨道占有率也将进一步促使其成为新的氢吸附活性位点。基于上述理论预测,间隙空位消除策略有望成为提高过渡金属氮化物HER性能的新手段。

图1. Mo2CN的结构模型和电子特性
传统过渡金属氮化物的合成方法多基于氧化物或金属的氨气退火氮化,间隙空位的改善只能依赖于杂原子的少量掺杂,电子结构调节程度十分有限。为解决上述问题,作者自主构建了一种聚三嗪配体锚定金属钼离子的纳米带结构,开发了原位调节碳、氮分压的制备手段,实现了无本征间隙空位的碳氮化钼晶体的构建。联合球差矫正电镜(Ac-HRTEM))和同步辐射(XANES、 EXAFS)等多种表征手段证明了材料的化学计量型晶体结构,配位结构和能带结构(图2, 3)。

图2. 碳氮化钼材料的晶体结构表征

图3. 碳氮化钼材料的配位结构表征
得益于本征间隙空位的消除,碳氮化钼催化剂的催化性能得到极大的提升,过电位、塔菲尔值分别降低至-84 mV和77.8 mv dec-1,均优于氮化钼、碳化钼等大部分已报道的钼基材料。高电流密度下的过电位也低于贵金属铂催化剂,表现出优异的碱性HER性能。同时,计时电位测试表明:材料经过14 h的催化测试,其过电位无明显上升,500次循环后的LSV曲线也没有明显变化,材料具有良好的催化稳定性。全解水测试系统则进一步证实了该化学计量型碳氮化钼材料高效、稳定的碱性HER性能(图4)。

图4. 碳氮化钼材料的碱性HER性能研究
基于上述实验结果,作者采用DFT理论计算和原位拉曼光谱揭示了化学计量型碳氮化钼的HER催化机理。DFT理论计算表明随着本征间隙空位的消除,材料的氢吸附自由能明显降低,晶格碳原子表现出最优氢吸附活性位点,与理论预测一致;原位拉曼光谱测试发现,在碱性HER测试过程中碳氮化钼表面出现明显的C-H键的拉曼振动,这进一步证实上述DFT计算结果。此外,作者基于理论计算对碳氮化钼表面的水分子吸附解离过程进行深度分析:随着晶格碳原子的引入,钼原子的氧化态上升,使其对水分子的氧端吸附增加,进而降低了水分子在材料表面的解离能,更有利于提供碱性HER的氢质子来源。综上所述,本征间隙空位的消除策略同时优化了水分子的吸附解离和氢物种的吸附,因而促进了碱性HER过程(图5)。
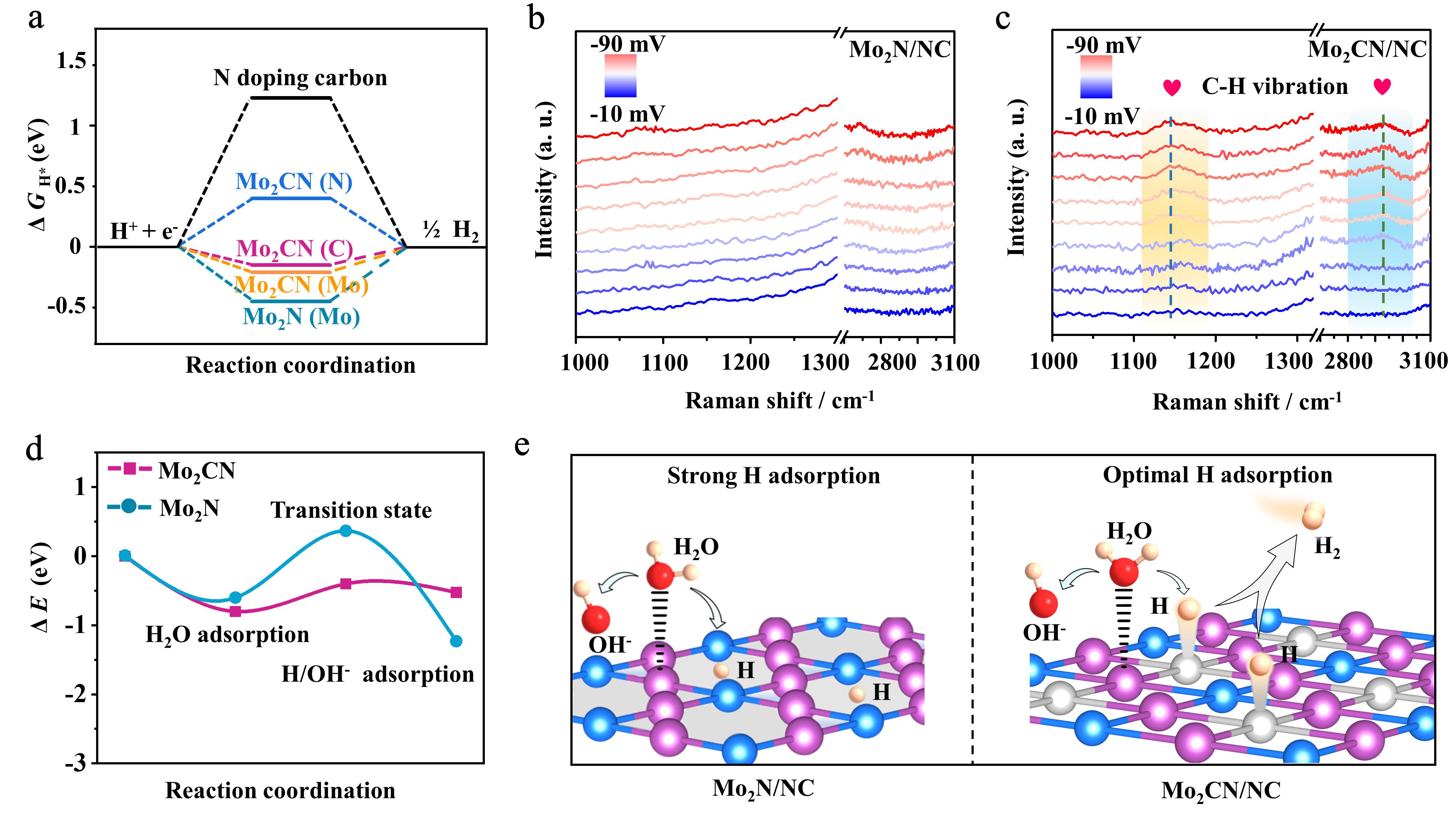
图5. 碳氮化钼材料的催化机理研究
该工作得到了国家自然科学基金和江苏省自然科学基金项目的支持。
声明:化学加刊发或者转载此文只是出于传递、分享更多信息之目的,并不意味认同其观点或证实其描述。若有来源标注错误或侵犯了您的合法权益,请作者持权属证明与本网联系,我们将及时更正、删除,谢谢。 电话:18676881059,邮箱:gongjian@huaxuejia.cn
